单细胞测序学习笔记(一)——细胞聚类和鉴定
使用的是10X Genomics
5k Peripheral blood mononuclear cells (PBMCs) from a healthy donor (v3 chemistry)
Single Cell Gene Expression Dataset by Cell Ranger 3.0.2
的数据
1.保持好习惯,先清空一下环境
rm(list = ls())2. 读取下载的数据,文件夹中包括三个文件:1. barcodes.tsv 2. features.tsv 3. matrix.mtx,将这三个文件加载到环境中。
pbmc.data <- Read10X(data.dir = "D:/myprojects/5k pbmc/5k_pbmc_v3_filtered_feature_bc_matrix/filtered_feature_bc_matrix")3. 创建Seurat对象,counts为读取的源文件,project为Seurat对象想保存的文件名,可以加上限定条件:min.cells为组织中分离的最少细胞数,min.features为一个细胞中测出的最少的基因数量
pbmc <- CreateSeuratObject(counts=pbmc.data,project = "pbmc5k",min.cells = 3,min.features = 200 )4. 查看一下pbmc的构成
> pbmc
An object of class Seurat
18791 features across 4962 samples within 1 assay
Active assay: RNA (18791 features, 0 variable features)5. 进行质控
创建一列名为percent.mt的新数据,添加到pbmc中,使用PercentageFeatureSet函数(此函数可以计算每个细胞中每一细胞器的QC指标)计算线粒体基因占比
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc,pattern = "^MT-")%>%为管道函数,就是把左件的值发送给右件的表达式(暂存在内存当中,没有保存为对象在硬盘中)
6. 查看一下
[email protected] %>% head()
orig.ident nCount_RNA nFeature_RNA percent.mt
AAACCCAAGCGTATGG-1 pbmc5k 13536 3502 10.7
AAACCCAGTCCTACAA-1 pbmc5k 12667 3380 5.6
AAACCCATCACCTCAC-1 pbmc5k 962 346 53.1
AAACGCTAGGGCATGT-1 pbmc5k 5788 1799 10.6
AAACGCTGTAGGTACG-1 pbmc5k 13185 2886 7.8
AAACGCTGTGTCCGGT-1 pbmc5k 15495 3801 7.57. 将QC结果展示为小提琴图,features中的名称加引号,ncol=3表示图形分三列展示
> VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)8. 可以结合散点图一起进行极值细胞的筛选删除
plot1 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(pbmc, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")9. 取极值删除后的子集
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 5000 & percent.mt < 25)删除后的小提琴图和点图


10. 标准化
通常情况下采用全局缩放的归一化方法"LogNormalize",新方法有“SCTransform” ,是一种三合一的方法,可以将质控,归一化和去识别高变基因合为一体
LogNormalize
> pbmc <- NormalizeData(pbmc, normalization.method = "LogNormalize", scale.factor = 10000)SCTransform
pbmc <- SCTransform(pbmc, vars.to.regress = "percent.mt", verbose = FALSE)
#随后保存
save(pbmc,file='filtered_gene_bc_matrices/hg19/01_pbmc3k_sctf.rd',compress=TRUE)11. 选择高变基因,使用“vst”方法选择2000个高变基因
pbmc <- FindVariableFeatures(pbmc, selection.method = "vst", nfeatures = 2000)12. 储存前10位高变基因
top10 <- head(VariableFeatures(pbmc), 10)
> plot3 <- VariableFeaturePlot(pbmc)#高变基因散点图
> plot4 <- LabelPoints(plot=plot3, points = top10,repel=TRUE)#top10加上基因名标签
> CombinePlots(plots = list(plot3, plot4), ncol =1)#结合到一张图中
13. Scale data
将均质为0标准差为1的数据记为标准化数据
> all.genes <- rownames(pbmc)
> pbmc <- ScaleData(pbmc,features = all.genes)这一步的目的主要是为了下一步PCA做准备
14. PCA
对scale后的数据进行PCA,注意嵌套
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc), verbose = FALSE)对降维后的数据进行绘图,注意:reduction选项,默认是UMAP,然后是tSNE,最后才是PCA
p1<- DimPlot(pbmc, reduction = "pca")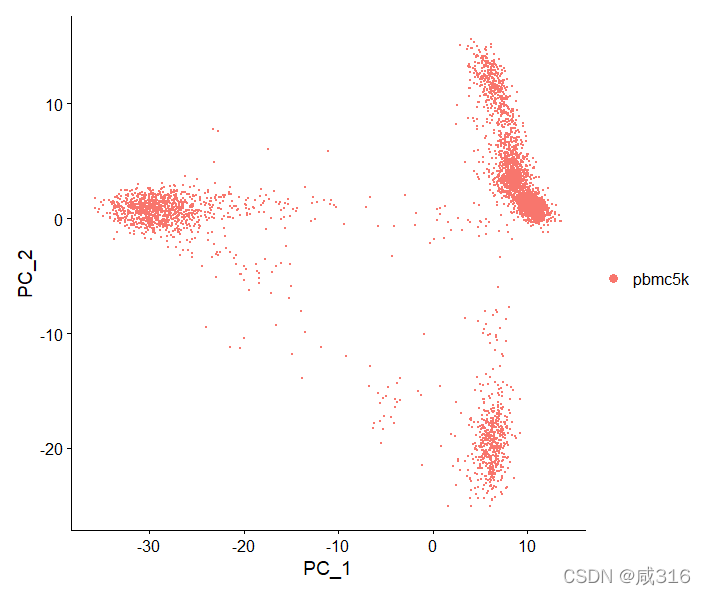
15. 检查PCA后的数据
方法多种,选一种即可
#Examine and visualize PCA results a few different ways
print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
VizDimLoadings(pbmc, dims = 1:2, reduction = "pca")
DimPlot(pbmc, reduction = "pca")
DimHeatmap(pbmc, dims = 1, cells = 500, balanced = TRUE)热图如下

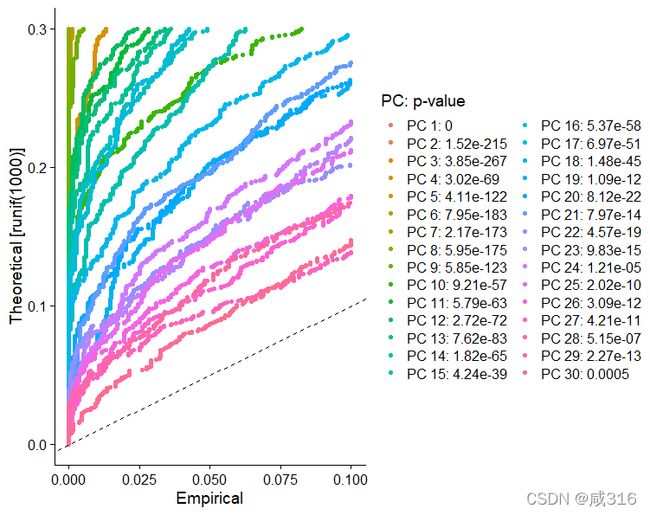
16. 在进行PCA后,我们要判断取多少个PC,判断方法有JackStraw和Elbow方法
> pbmc <- JackStraw(pbmc, num.replicate = 100, dims = 50)
> pbmc <- ScoreJackStraw(pbmc, dims = 1:50)
> JackStrawPlot(pbmc, dims = 1:30)这一步比较慢,这个数据集花了8 min左右
Elbow
ElbowPlot(pbmc, ndims = 50)
主要观察“胳膊肘”在哪里
17. 细胞分类
> pbmc <- FindNeighbors(pbmc, dims = 1:20)
> pbmc <- FindClusters(pbmc, resolution = 0.5)
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4595
Number of edges: 165447
Running Louvain algorithm...
0% 10 20 30 40 50 60 70 80 90 100%
[----|----|----|----|----|----|----|----|----|----|
**************************************************|
Maximum modularity in 10 random starts: 0.8981
Number of communities: 13
Elapsed time: 0 seconds这里设置了dims = 1:20 即选取前20个主成分来分类细胞。分类的结果如下,可以看到,细胞被分为13个类别。
选择不同的resolution值可以获得不同的cluster数目,值越大cluster数目越多,默认值是0.5.
Look at cluster IDs of the first 5 cells
> head(Idents(pbmc), 5)
AAACCCAAGCGTATGG-1 AAACCCAGTCCTACAA-1 AAACGCTAGGGCATGT-1 AAACGCTGTAGGTACG-1 AAACGCTGTGTCCGGT-1
1 1 5 0 1
Levels: 0 1 2 3 4 5 6 7 8 9 10 11 12接下来使用UMAP或者tSNE来进行细胞类别可视化
pbmc <- RunUMAP(pbmc, dims = 1:20)
pbmc <- RunTSNE(pbmc, dims = 1:20)
DimPlot(pbmc, reduction = "tsne")
DimPlot(pbmc, reduction = "UMAP", label=TRUE)

PCA可视化结果重叠度太高

保存一下
> saveRDS(pbmc, file = "pbmc5k_round1.rds")18.提取各个细胞类型的marker gene其中ident.1参数设置待分析的细胞类别,
min.pct表示该基因表达数目占该类细胞总数的比例
find all markers of cluster 1
cluster1.markers <- FindMarkers(pbmc, ident.1 = 1, min.pct = 0.25)
head(cluster1.markers, n = 5)
p_val avg_log2FC pct.1 pct.2 p_val_adj
RBP7 0 1.5 0.70 0.017 0
PGD 0 1.4 0.82 0.111 0
AGTRAP 0 1.5 0.90 0.233 0
TNFRSF1B 0 1.5 0.94 0.315 0
CDA 0 1.4 0.75 0.026 0find all markers distinguishing cluster 5 from clusters 0 and 3,寻找能将5号细胞从0和3号细胞中区别出来的markers
cluster5.markers <- FindMarkers(pbmc, ident.1 = 5, ident.2 = c(0, 3), min.pct = 0.25)
head(cluster5.markers, n = 5)
p_val avg_log2FC pct.1 pct.2 p_val_adj
ARHGAP24 0 1.7 0.81 0.002 0
BANK1 0 2.9 0.98 0.006 0
MARCH1 0 1.8 0.84 0.007 0
HLA-DQA1 0 3.6 0.99 0.012 0
HLA-DQB1 0 3.1 0.98 0.017 0# find markers for every cluster compared to all remaining cells, report only the positive ones找到与所有剩余细胞相比的每个类别细胞的marker(positive marker)
pbmc.markers <- FindAllMarkers(pbmc, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
top3 <- pbmc.markers %>% group_by(cluster) %>% top_n(n = 3, wt = avg_log2FC)由于要一个个计算,过程比较耗时
储存每类细胞marker的前三名,然后做热图可视化
DoHeatmap(pbmc, features = top3$gene) + NoLegend()
19. 下面,对marker genes进行可视化,可以直观的看到不同类别细胞与marker的联系
VlnPlot(pbmc, features = c("MS4A1", "CD79A"))
可以看出MS4A1在第5,6类细胞中高表达,CD79A在5,6高表达,8,12中表达但不高
也可以使用raw counts进行绘图
VlnPlot(pbmc, features = c("MS4A1", "CD79A"), slot = "counts", log = TRUE)
#slot="" Use non-normalized counts data for plotting使用非标准化的数据
20. 将marker基因的表达映射到细胞中(FeaturePlot)
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP", "CD8A"))UMAP聚类

tSNE聚类
FeaturePlot(pbmc, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A",
+ "FCGR3A", "LYZ", "PPBP", "CD8A"),reduction="tsne")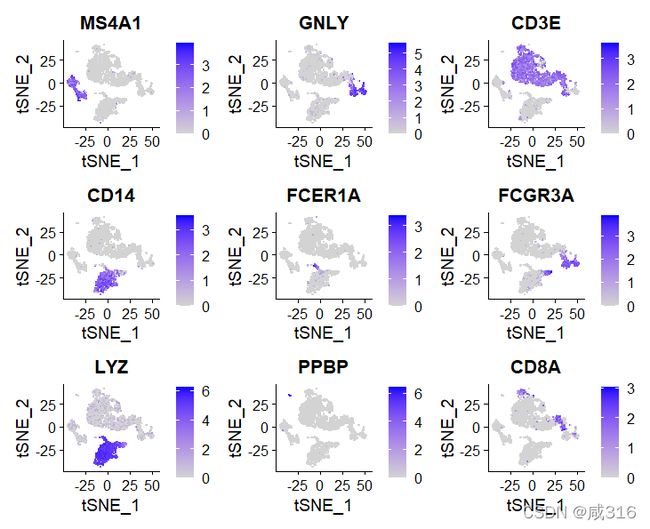
至此,我们可以通过先验知识来判断每个类别的细胞是什么细胞,根据已经发表的细胞特异性marker来判断