用Python进行GEO数据挖掘(学习笔记三):利用rpy2库调用R的limma包进行差异表达分析
加载需要的python库
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import rpy2
from rpy2.robjects import r
from rpy2.robjects.packages import importr
from rpy2.robjects import pandas2ri
pandas2ri.activate()1.数据准备
利用limma包做差异表达分析需要三个数据:表达矩阵、样本分组矩阵、差异比较矩阵
差异比较矩阵:即design。 差异比较矩阵就是告诉差异分析函数是要从要分析哪些变量间的差异,简单说就是说明哪些是对照哪些是处理。
导入已有数据:表达矩阵和样本分组信息
exp=pd.read_excel("F:\\bioinformatics\\20220806\\data\\GSE5281\\NEW2_GSE5281.xlsx",index_col='symbol')
exp.head()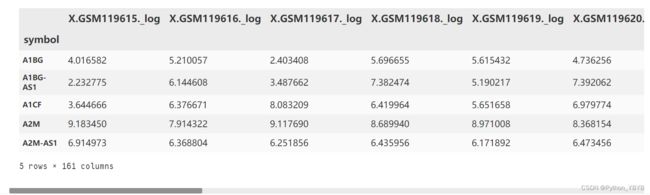
group_inf=pd.read_excel("F:\\bioinformatics\\20220806\\data\\GSE5281\\group_inf.xlsx",index_col=0)
group_inf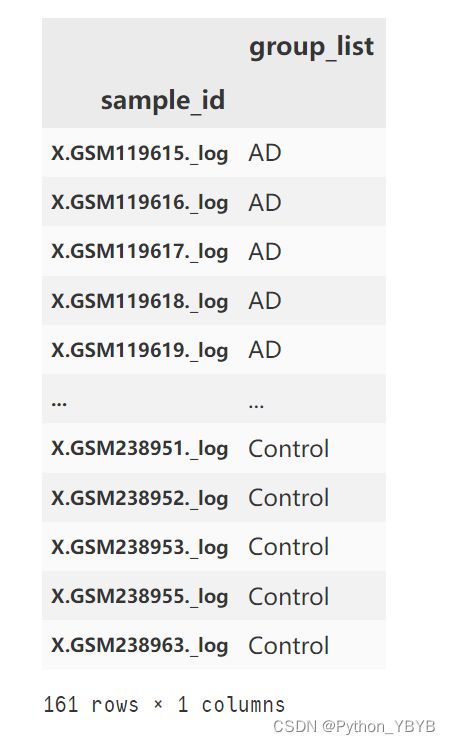
表达矩阵就是exp,下面用样本分组信息来做分组矩阵
a=group_inf['group_list'].tolist()
list1=[] ##存放AD的状态向量
list2=[] ##存放Control的状态向量
for i in a:
if i=="AD":
x=1
y=0
else:
x=0
y=1
list1.append(x)
list2.append(y)
design=pd.DataFrame({'AD':list1,"Control":list2},index=a)
design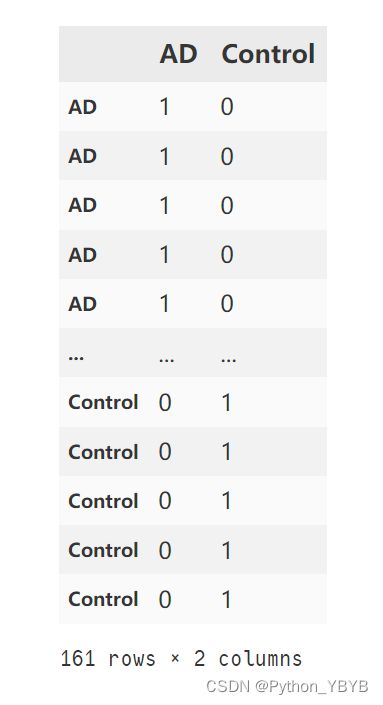
这个样子,分组矩阵design就得到了
还有一个矩阵,差异比较矩阵
limma=importr("limma")
contrast_matrix=limma.makeContrasts("AD-Control",levels=design)
contrast_matrix运行出来是这样的东西

-1和1的意思是,AD是用来比的,Control是用来被比的,即AD/Control
开始用limma包做差异分析
##step1
fit=limma.lmFit(exp,design)
##step2
fit2=limma.contrasts_fit(fit,contrast_matrix)
fit2=limma.eBayes(fit2)
##step3
DEG_ot=limma.topTable(fit2,adjust="fdr",coef=1,n=float("inf"))
print(DEG_ot) 运行结果部分截图
运行结果部分截图
发现好像不对,少了两列,什么原因现在我还没有搞明白,大家也可以指教一下这个地方。无奈只好调用R脚本文件来运行代码。
2.limma包进行差异表达分析
将差异比较矩阵的生成包含在内进行:
limma=importr("limma")
##数据转换
rdf1=pandas2ri.py2rpy(exp)
rdf2=pandas2ri.py2rpy(design)
from rpy2.robjects import globalenv ##让globalenv来激活rdf
globalenv['rdf1']=rdf1
globalenv['rdf2']=rdf2
rscript='''
contrasts_matrix<-makeContrasts("AD-Control",levels=rdf2)
fit<-lmFit(rdf1,rdf2)
fit2<-contrasts.fit(fit,contrasts_matrix)
fit2<-eBayes(fit2)
temp<-topTable(fit2,coef=1,n=Inf)
nrDEG=na.omit(temp)
'''
r(rscript)
这下结果就出来了!
后面调用这个结果,就直接调用r(rscript)就行了,它是一个DataFrame数据框
3.差异基因的筛选以及可视化
(1)提取差异表达基因
从差异分析的输出结果中可以看到,其中包含转录本名称,log2FC以及adjusted等等数据。差异基因的筛选主要看log2FC以及adjusted或unajusted的P值。 log2FC的默认cut-off值是 > | 2 |; P值的默认cut-off值为10e-6。在这里可以设置阈值: log2FC:1,pvalue:0.05
result=r(rscript) ##赋值给新的变量,方便后面调用
fold_cutoff=1
adjp_cutoff=0.05
list3=[] #准备一个列表来装接下来差异条件遍历的结果
normal_list=[] ##装没有显著差异的symbol ID
for j in range(0,len(result.index)):
if (abs(result['logFC'][j])>=fold_cutoff) and (result['adj.P.Val'][j]<=adjp_cutoff):
list3.append(j)
else:
normal_list.append(result.index.tolist()[j])
print("差异表达基因的数量:",len(list3))
print("差异不显著基因的数量:",len(normal_list))

发现有727个差异表达基因,下面就看看上调和下调的差异表达基因数量:
up_list=[] #存放上调基因symbol ID
for h1 in range(0,len(result.index)):
if (result['logFC'][h1]>=fold_cutoff) and (result['adj.P.Val'][h1]<=adjp_cutoff):
up_list.append(result.index.tolist()[h1])
down_list=[] ##存放下调基因symbol ID
for h2 in range(0,len(result.index)):
if (result['logFC'][h2]<-(fold_cutoff)) and (result['adj.P.Val'][h2]<=adjp_cutoff):
down_list.append(result.index.tolist()[h2])
print("上调基因数量:",len(up_list))
print("下调基因数量:",len(down_list))
print("差异表达基因数量:",len(up_list)+len(down_list))
下面提取上调和下调的差异表达基因
DEG_up=exp[exp.index.isin(up_list)]
DEG_down=exp[exp.index.isin(down_list)]
print("上调基因的维数:",DEG_up.shape)
print("下调基因的维数",DEG_down.shape)
DEG_up['up/down']=['up']*275 #添加标签
DEG_down['up/down']=['down']*452 #添加标签
DEG_matrix=pd.concat([DEG_up,DEG_down],join='inner') ##合并
DEG_matrix
DEG_matrix.to_excel("F:\\bioinformatics\\20220806\\data\\GSE5281\\DEG_GSE5281.xlsx",index=True) #保存下来
这个样子,差异表达基因就得到了!
(2)差异表达分析可视化
先做画图之前的一些准备
result['adj.P.Val_log']=-np.log10(result['adj.P.Val']) #将adj.P.Val列的值全部负对数转换
result['sig']='normal' #新设一列sig全部为normal
result.loc[(result['logFC']>=1)&(result['adj.P.Val']<0.05),'sig']='up' #上调基因sig将normal替换为up
result.loc[(result['logFC']<-1)&(result['adj.P.Val']<0.05),'sig']='down' ##上调基因sig将normal替换为down
result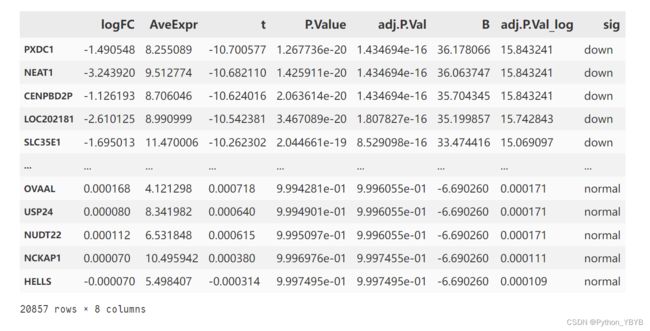
开始绘制
import seaborn as sns #导入seaborn来画
##先设置一下颜色
colors=["#01c5c4","#ff414d","#686d76"]
sns.set_palette(sns.color_palette(colors))
#绘图
ax=sns.scatterplot(x='logFC',y='adj.P.Val_log',data=result,
hue='sig',#颜色映射
edgecolor=None,#点边界颜色
s=8,#点大小
)
#标签
ax.set_title("DEG vocalno")
ax.set_xlabel("logFC")
ax.set_ylabel("-log10(adj.P.Val)")
#图例
ax.legend(loc='center right',bbox_to_anchor=(0.95,0.76),ncol=1)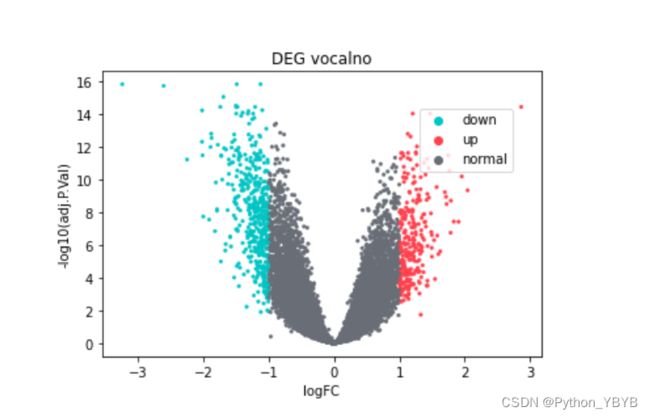
##绘图:聚类热图
DEG_matrix.drop(columns='up/down')
sns.clustermap(DEG_matrix,cmap='afmhot', standard_scale = 0)
从图中就可以看出,上部分基因在第一组中高表达,在第二组中低表达,下半部分基因在第一组中低表达,在二组中高表达。
接下来就可以用差异基因做各种分析啦!