2022.04.14【读书笔记】|WGCNA分析原理和数据挖掘技巧
文章目录
- 背景
-
- RNA-seq分析一般逻辑
- 局限性
- 基因表达和调控方式
- 与网络相关的基础概念
-
- 无尺度网络
- 模块以及模块特征值
- 连通性
-
- 非权重网络
- 权重网络
- 核心基因
- WGCNA网络原理和构建过程
-
- 简介
- 哲学理念
- 网络构建的两个核心步骤
-
- 基因的过滤
-
- 去除低丰度的基因
- 去除没有变化的基因
- 最终期望:
- 一个务实的策略——双重标准:
- 计算基因间的相关性
-
- 基因相关关系的无尺度化
- 关键参数β的确定
- 基因间表达调控的相关性
- 将基因划分为模块
- WGCNA网络生物学意义的挖掘
-
- 目标模块选取
-
- 模块表达模式分析
- 模块与其他指标的相关性分析
- 富集分析(常见为功能富集分析)
- 从目标基因直接入手
- 模块内的分析
-
- 模块内连通性(Module connectivity)
- 核心基因分析
- 目标基因相关的局部调控网络
- 关注特定类型的基因
- 总结
-
- 工具推荐
- 样品要求
- 步骤
- 特点
背景
RNA-seq分析一般逻辑
1.上下调
2.趋势分析
对分类基因开展功能、调控分析
局限性
大样本情况,无法进行有效分类
依赖数据的功能分析无法推测新的调控关系
基因表达和调控方式
1、组成型表达(constitutive gene expression)
细胞中持续进行的基因表达,且较少受环境因素的影响。这类基因通常被称为持家基因(housekeeping gene)。
2、基因间相互诱导和阻遏表达
即一个基因诱导(例如转录因子)或阻遏(例如 miRNA)另外一个基因的转录翻译。
3、协调表达
在一定机制控制下,功能相关的一组基因,协调一致,共同表达。
与网络相关的基础概念
网络图的两大元素:
• 点:图中每一个圆圈代表一个节点,如基因。
• 边:在基因调控网络中,基因相互间的调控关系构成了边。
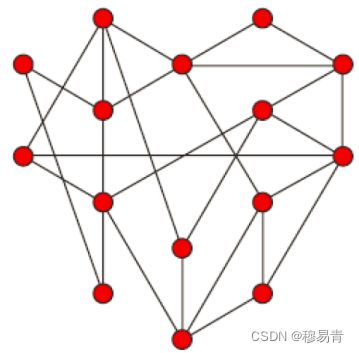
无尺度网络
无尺度网络:大部分节点只和很少节点连接,而有极少的节点与非常多的
节点连接。
数学上的描述:假设m为某节点的连接数。统计所有基因的m值,然后以
m高低为指标对所有基因分类。n为节点连接数为m的基因的数量。m与n
应该成反比。

模块以及模块特征值
模块(module)
表达模式相似的一组基因。趋势分析的本质也是分模块,但其功能没有WGCNA中的方法强大。
模块特征值(module eigengene)
模块中的所有基因进行PCA分析,得到的主成分1(PC1)的值。PC1相当于模块中所有基因表达量的加权,可以代表这个模块的表达模式。
连通性
一个基因与其他基因的连接程度(通常只在模块内计算)。常称为
connectivity或degree,或用数字k表示。有两种计算方法:
非权重网络
当两个基因的相关性大于某个值(例如0.6),才认为有相关性。最后某个基因的k值等于与其相关的基因的数量。红圈中的基因k=3。

权重网络
所有两两基因的相关性都被保留(无论强弱)。某个基因的k值等于其与各个基因的相关性之和。红圈中的基因k=四个相关性之和。

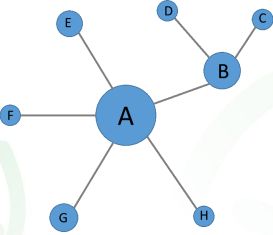
核心基因
在一个模块中,连通性(k值)排名靠前的基因。K值排名靠前本身已
经表明它们处于中心枢纽的位置。如下图中的A基因。
WGCNA网络原理和构建过程
简介
WGCNA,全称weighted gene co-expression network analysis,即权重基因共表达网络分析。自2005年B Zhang, S Horvath等提出,至今已被引用2268次。在疾病以及其他性状与基因关联分析等方面的研究中被广泛应用。
哲学理念
从“系统”的角度去解析关注的问题,而不是去罗列单一基因的清单。
– 描述整个“发动机”的工作原理,而不是去摆放所有的螺丝钉。
• 研究的关注点是基因模块,而不是单一基因。
• 网络的概念让抽象的生物学问题更直观易懂。
网络构建的两个核心步骤
基因的过滤
基因的过滤并没有统一标准;
不要使用所有基因,原因:1)计算慢;2)上限是4万;3)一些基因没有意义,例如低丰度、没有变化的基因等;
一般的基因过滤标准:
去除低丰度的基因
范例举例:
表达量在所有样本均小于1的基因;
表达量在 x % 样本=0的基因,且最大值小于1。
注意,要具体问题具体分析。例如,50%样本 =0 这个标准,对随机群体
可能是合适的。但如果样本是病例组:对照组,那么50% 样本 = 0 则可能是非常有意义的基因。
去除没有变化的基因
在样本数巨大情况下,差异基因并集策略是没用的(因为等于没过滤)
采取计算变异系数,然后过滤掉最低的x% 是更适用的策略
最终期望:
基因数量在几千~2万以内;
要包含老师关注的基因(的确,很多情况老师一开始不知道自己要什么,发现没有后,要求重新过滤分析)
一个务实的策略——双重标准:
按照一定标准过滤;
然后加入老师潜在关注的基因(例如可以提前提供,或不可避免重新分析的时候加入);
计算基因间的相关性
基因相关关系的无尺度化
处理过程
1)原始S矩阵:Sij=|cor (xi, xj)| # 计算基因间两两相关性
2) 无尺度化(拉大贫富差距),确定最佳β值,得到A矩阵
A矩阵: aij=power (Sij, β)=|Smn|β
注:在WGCNA中,应该只考虑正相关的基因。幂函数处理的作用:
a)在幂函数处理后,少量强相关性的关系(例如:r2=0.999)不受影响或影响较少,相关性弱的关系(例如:r2=0.1)取n次幂后,相关性下降明显。
b) 最后导致网络关系无尺度化:强相关的关系少,弱相关的关系多;
连通性高的基因少,连通性弱的基因多。即,满足上文提到的无尺度网络的定义“假设m为某节点的连接数。统计所有基因的m值,然后以m高低为指标对所有基因分类。n为节点连接数为m的基因的数量。
m与n应该成反比。
备注: 基因i的连通性: 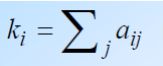
即其与其它基因的相关性之和。
关键参数β的确定

β值:WGCNA分析的第一个关键参数。
左图:不同β值下,m与n的相关性的变化(上一页提到,理论上是负值,应该做过转换)。一般认为取β值大于0.8或到达平台期时最小的β值用于构建网络。
右图:不同β值下,所有基因连通性的均值。
β值:但不应该死抱β值大于 0.8这个标准。
(1)数据由于噪音等其他因素,可能会β值早早到达平台期(例如 0.6),那么我们应该接受这个情况;
遇到过一些数据噪音极大的情况,β值上下剧烈波动;
过滤掉一个低丰度的基因,是个值得尝试的策略;
(2)β值不宜取太大,例如 大于 20;
基因间表达调控的相关性
3)如何评估两个基因的表达模式的相关性
生物学逻辑:调控的相关性=直接相关 + 间接相关
数学实现过程:基于A矩阵,计算两两基因间的TOM值
详析TOM值 :TOM值= 直接相关 + 间接相关(强化直接相关的值)
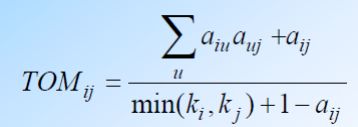
将基因划分为模块
层级聚类树与区分模块

聚类和分模块的几个关键参数

调控网络中的各个定义
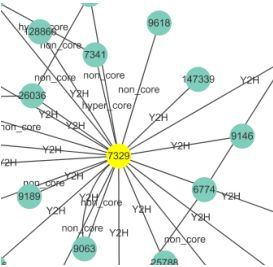
点和边的定义已经介绍过;
Degree(度),又称为连通性(Connectivity),即1个基因拥有的边(调控关系)的数量;
注:
1)WGCNA中,连通性是各个边的相关系数(0~1)之和;
2)cytocape软件中有1个定义——Neighborhoodconnectivity指的是相邻基因连通性,而不是这个基因本身;
Hub gene:在1个调控网络中,位于中心的基因,即连通性排名靠前的基因。
WGCNA网络生物学意义的挖掘
目标模块选取
模块表达模式分析
模块特征值(即PC1)在各个样本中的丰度,构成了这个模块的表达模式/规律

模块与其他指标的相关性分析
1个模块相当于代表一类基因,模块的特征值可以某种程度上代表一类基因的表达模式
样本在各个模块特征值可以和样本的性状开展相关分析,找出与特定性状相关联的模块
模块也可以其他信息开展相关性分析,例如 样本SNP基因型,样本分组信息关于样本的相关性:对分析与某类样本的相关性的时候,可以将目标类型定义为1,其他类型定义为0.

富集分析(常见为功能富集分析)
如上文提到的 存在诱导/阻遏表达(TF和靶基因)或协调表达(例如被同一个TF调控一组基因)关系的一组基因,更容易出现在一个模块中。而且在大样本的情况下,基因的表达分类更加有规律。
所以,对模块开展KEGG、GO功能富集分析,通常会找到很有规律的功能类型。
从目标基因直接入手
当然,如果有目标基因,也可以直接找其所在的模块,然后进行进行下一步分析——模块内的功能调控关系分析
模块内的分析
模块内连通性(Module connectivity)
WGCNA模块内输出的Connectivity是模块内的连通性,是这个基因与其他基因相关性之和(记住:是aij,而不是TOM之和),所以这是软阈值(soft threshold)的计算方法(请回顾上文)。
在某些文章里,也会使用硬阈值(hard threshold)的方法,即认为TOM值(就是模块调控关系表中的weight值)大于X(默认是0.15)的两个基因才认为是相关的,然后计算每个基因的连接数量(可以先过滤有足够强度的关系,然后导入cytoscape或OS-tools计算)
我们比较推荐使用前者。
核心基因分析
模块中连通性较高的基因(例如人为设定排名前30或前10%),被称为hub基因。
高连通性的Hub基因通常为调控因子(调控网络中处于偏上游的位置),而低连通性的基因通常为调控网络中偏下游的基因(例如,转运蛋白、催化酶等)
我们应该按照自己研究目的,充分利用连通性这个信息。
目标基因相关的局部调控网络
从目标基因入手,找与之TOM值排名靠前(例如前10)或TOM值大于某个阈值的基因列表。通过这一策略,可以准确筛选潜在与目标基因存在调控关系的候选基因,这些基因是下阶段功能验证的优先候选。

从目标基因入手,找与之TOM值排名靠前(例如前10)或TOM值大于某个阈值的基因列表。后者是更稳定的策略。因为如果目标基因不是Hub基因(而是下游的基因),那么与其TOM值大于0.15或0.3 的基因是非常少的。因为间接相关的基因太少,与其相关的TOM值不会太大。

使用多类信心进行筛选:
1)如果目标基因是下游基因,则可以找共同的上游调控基因;
2)如果目标基因是上游基因或hub基因,则可以挖掘共同调控的下游基因;
3)结合转录因子等其他注释信息,也有利于解读。

绘制策略
1)确定目标基因(这里是3个)
2)筛选与每个目标基因TOM值排名前n的其他基因(这里取n=10)
3)图形中的调控关系,只画目标基因与其他基因的关系,但不画其他
基因与其他基因的关系,避免图形过滤冗杂
关注特定类型的基因
可以结合基因注释信息,关注模块内特定类型的基因,例如转录因子。
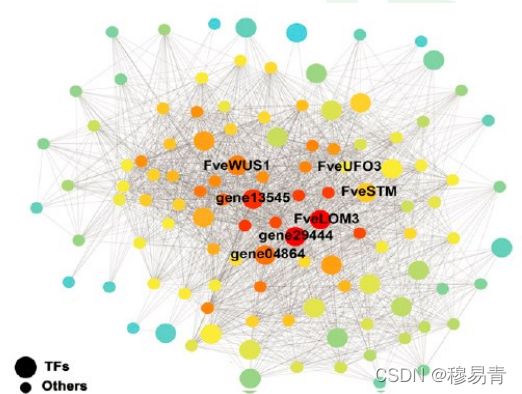
以上的大部分图形(除网络图),都是WGCNA R包可以输出。
• 网络图的必须自己定制:
(1)筛选目标基因(核心基因,已知目标基因)
(2) 筛选与目标基因相关的调控关系/基因(TOM值过滤)
(3) 整合多种信息,如目标调控关系、TOM值(调控强度)、连通性、基因种类(如转录因子),结合绘图软件(cytoscape或 OS-tools网络图工具),就可以将以上得到的信息图形化。
总结
工具推荐
本地版Cytoscape教程:
第19期在线交流cytoscape介绍与实操【视频】
OS-tools 在线版cytoscape(做过优化,更适应cytoscape的原始输出结果和简单的数据过滤)
样品要求
- WGCNA分析对样本有什么要求?
答:
我们推荐以下的样品数:
1)不含生物学重复的独立样本组:样本数≥8
2)包含生物学重复的样本组:样本数≥15
主要考虑的因素:
1)样本必须包含丰富的变化信息,才能区分为多个有意义的生物学模块(需要多个独立处理组)。
2)必须保证有多个样本,才能保证相关系数计算的准确性。
步骤
关系矩阵构建
基本原理: 利用基因间表达量的相关系数。
模块识别
基本原理:利用拓扑树结构区分基因模块。
核心模块挑选
基本原理:分析模块内基因的特性,进一步寻找有生物学意义的模块。
分析策略:模块特征值与表型间的相关性,模块内基因的KO、GO分析。
核心基因的挑选,并构建网络
基本原理:利用基因连通性信息挑选核心基因,并围绕其构建网络
特点
WGCNA 是一种系统的遗传分析方法,十分适用于分析
复杂性状/疾病:
1)基于RNA表达量的调控关系分析,不需要基因间作用关系的先验信息。
2)强调模块 (通路),而不是单一基因。
精简了组学水平的海量信息
在与其他信息(如表型)的相关性分析中,减少了多重检验校正的影响。
注:几十个模块的相关性分析 vs 几千个基因的相关性分析
3)使用基因连通性便于找到核心基因
核心基因在功能实验中往往有更高验证率。
4)调控网络相对稳定,受样本量影响小
相关调控网络基于相关系数,而不是p value(p value受样本量影响大)。因此,保证来源不同,样本数不同的数据间的可比较性