【生信学习】Scanpy函数pp.calculate_qc_metrics
一、学习一个新的函数
通过help该函数,可以得到一个简单的参考提示。

但如果想要更深一步的去学习函数的源文件。那么就把光标停留在函数上几秒,就能找到源文件的文件目录。我的源文件目录是:
D:\Anaconda\Lib\site-packages\scanpy\preprocessing\_qc.py
二、主要代码
def calculate_qc_metrics(
adata: AnnData,
*,
expr_type: str = "counts",
var_type: str = "genes",
qc_vars: Collection[str] = (),
percent_top: Optional[Collection[int]] = (50, 100, 200, 500),
layer: Optional[str] = None,
use_raw: bool = False,
inplace: bool = False,
log1p: bool = True,
parallel: Optional[bool] = None,
) -> Optional[Tuple[pd.DataFrame, pd.DataFrame]]:
X = _choose_mtx_rep(adata, use_raw, layer)
obs_metrics = describe_obs(
adata,
expr_type=expr_type,
var_type=var_type,
qc_vars=qc_vars,
percent_top=percent_top,
inplace=inplace,
X=X,
log1p=log1p,
)
var_metrics = describe_var(
adata,
expr_type=expr_type,
var_type=var_type,
inplace=inplace,
X=X,
log1p=log1p,
)
if not inplace:
return obs_metrics, var_metrics
上面的代码中调用了两个函数describe_obs, describe_var,来进行对细胞、基因进行质控指标的计算。在这里有个比较重要的参数inplace,影响着结果的查看。
1、inplace = False
会直接返回细胞、基因两个质控指标。obs对应的是细胞,var对应的是基因。这个跟Anndata数据格式相关。
以一个单细胞分析作为展示,数据集包括有2041个细胞,1020个基因。
sc.pp.calculate_qc_metrics(adata, percent_top=None, inplace=False) #返回控制指标
2、inplace = True
如果inplace=True,会将质控指标添加到adata.obs,adata.var中。输出adata.var展示查看一下。
sc.pp.calculate_qc_metrics(adata, percent_top=None, inplace=True)
adata.var
三、参数详解
1、参数概况
(1)obs
n_genes_by_counts: 每个细胞中有多少种基因
total_counts: 该细胞的基因总数
(2)var
n_cells_by_counts: 所有细胞中表达该基因的细胞数目
mean_counts: 所有细胞中该基因表达的平均值
pct_dropout_by_counts: 未表达该基因的细胞占细胞总数的百分比
total_counts: 所有细胞中,基因的表达量总和
2、describe_obs
def describe_obs(
adata: AnnData,
*,
expr_type: str = "counts",
var_type: str = "genes",
qc_vars: Collection[str] = (),
percent_top: Optional[Collection[int]] = (50, 100, 200, 500),
layer: Optional[str] = None,
use_raw: bool = False,
log1p: Optional[str] = True,
inplace: bool = False,
X=None,
) -> Optional[pd.DataFrame]:
# Handle whether X is passed
if X is None:
X = _choose_mtx_rep(adata, use_raw, layer)
if isspmatrix_coo(X):
X = csr_matrix(X) # COO not subscriptable
if issparse(X):
X.eliminate_zeros()
obs_metrics = pd.DataFrame(index=adata.obs_names)
if issparse(X):
obs_metrics[f"n_{var_type}_by_{expr_type}"] = X.getnnz(axis=1)
else:
obs_metrics[f"n_{var_type}_by_{expr_type}"] = np.count_nonzero(X, axis=1)
if log1p:
obs_metrics[f"log1p_n_{var_type}_by_{expr_type}"] = np.log1p(
obs_metrics[f"n_{var_type}_by_{expr_type}"]
)
obs_metrics[f"total_{expr_type}"] = np.ravel(X.sum(axis=1))
if log1p:
obs_metrics[f"log1p_total_{expr_type}"] = np.log1p(
obs_metrics[f"total_{expr_type}"]
)
if percent_top:
percent_top = sorted(percent_top)
proportions = top_segment_proportions(X, percent_top)
for i, n in enumerate(percent_top):
obs_metrics[f"pct_{expr_type}_in_top_{n}_{var_type}"] = (
proportions[:, i] * 100
)
for qc_var in qc_vars:
obs_metrics[f"total_{expr_type}_{qc_var}"] = np.ravel(
X[:, adata.var[qc_var].values].sum(axis=1)
)
if log1p:
obs_metrics[f"log1p_total_{expr_type}_{qc_var}"] = np.log1p(
obs_metrics[f"total_{expr_type}_{qc_var}"]
)
obs_metrics[f"pct_{expr_type}_{qc_var}"] = (
obs_metrics[f"total_{expr_type}_{qc_var}"]
/ obs_metrics[f"total_{expr_type}"]
* 100
)
if inplace:
adata.obs[obs_metrics.columns] = obs_metrics
else:
return obs_metrics
(1)处理参数
如果没有传递参数,那么在adata中重新获取。

(2)obs文件生成DataFrame
obs_metrics = pd.DataFrame(index=adata.obs_names)其中行名为细胞的名称,生成的列包括有:
n_genes_by_counts, log1p_n_genes_by_counts, total_counts, log1p_total_counts.
(3)n_genes_by_counts
n_genes_by_counts:每个细胞中基因的基因种类。通俗一点讲:一个细胞中有多少种基因。

np.count_nonzero(X, axis = 1)计算了每行细胞中表达量非0的基因的数量。
(4)log1p_n_genes_by_counts
log1p_n_genes_by_counts 表示对n_genes_by_counts进行log1p处理,log1p= log(X+1)。默认log1p=True, 即进行转换处理。

(5)total_counts
该细胞中的基因总数
obs_metrics[f"total_{expr_type}"] = np.ravel(X.sum(axis=1))(6)log1p_total_counts
log1p_total_counts 表示对total_counts进行log1p处理,log1p= log(X+1)。默认log1p=True, 即进行转换处理。

(7)pct_counts_in_top_{n}_genes

percent_top默认值为(50,100,200,500) ,该参数用于计算每个细胞中前n个基因的表达量和这n个基因的表达量占总基因表达量的比例。
函数top_segment_proportions用于计算这个比例。for循环将percent_top中每个n值,所计算的比例转换成百分比,并保存在obs_metrics 这个DataFrame中。
(8)qc_vars 相关指标计算
qc_vars 用于指定adata.var里的特定字段。
adata.var有个字段为"mt" 用于判断基因是否为线粒体基因,将会增加三个指标:
total_counts_mt : 细胞中线粒体基因表达量总和
log1p_total_counts_mt: log1p(细胞中线粒体基因表达量总和)
pct_counts_mt: 细胞中线粒体基因表达量总和 占 总基因表达和的百分比

2、describe_var
def describe_var(
adata: AnnData,
*,
expr_type: str = "counts",
var_type: str = "genes",
layer: Optional[str] = None,
use_raw: bool = False,
inplace=False,
log1p=True,
X=None,
) -> Optional[pd.DataFrame]:
# Handle whether X is passed
if X is None:
X = _choose_mtx_rep(adata, use_raw, layer)
if isspmatrix_coo(X):
X = csr_matrix(X) # COO not subscriptable
if issparse(X):
X.eliminate_zeros()
var_metrics = pd.DataFrame(index=adata.var_names)
if issparse(X):
# Current memory bottleneck for csr matrices:
var_metrics["n_cells_by_{expr_type}"] = X.getnnz(axis=0)
var_metrics["mean_{expr_type}"] = mean_variance_axis(X, axis=0)[0]
else:
var_metrics["n_cells_by_{expr_type}"] = np.count_nonzero(X, axis=0)
var_metrics["mean_{expr_type}"] = X.mean(axis=0)
if log1p:
var_metrics["log1p_mean_{expr_type}"] = np.log1p(
var_metrics["mean_{expr_type}"]
)
var_metrics["pct_dropout_by_{expr_type}"] = (
1 - var_metrics["n_cells_by_{expr_type}"] / X.shape[0]
) * 100
var_metrics["total_{expr_type}"] = np.ravel(X.sum(axis=0))
if log1p:
var_metrics["log1p_total_{expr_type}"] = np.log1p(
var_metrics["total_{expr_type}"]
)
# Relabel
new_colnames = []
for col in var_metrics.columns:
new_colnames.append(col.format(**locals()))
var_metrics.columns = new_colnames
if inplace:
adata.var[var_metrics.columns] = var_metrics
else:
return var_metrics
(1)处理参数
如果没有传递参数,那么在adata中重新获取。

(2)obs文件生成DataFrame
var_metrics = pd.DataFrame(index=adata.var_names)其中行名为基因的名称,生成的列包括有:
n_cells_by_counts, mean_counts, log1p_mean_counts, pct_dropout_by_counts, total_counts, log1p_total_counts。

(3)n_cells_by_counts和mean_counts

n_cells_by_counts: 所有细胞中表达该基因的细胞数目
mean_counts: 所有细胞中该基因表达的平均值
(4)log1p_mean_counts
log(mean_counts+1)

(5)pct_dropout_by_counts
未表达该基因的细胞占细胞总数的百分比。
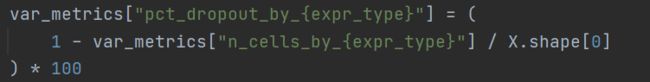
(6)total_counts
所有细胞中,基因的表达量总和。
var_metrics["total_{expr_type}"] = np.ravel(X.sum(axis=0))四、参考
1、Scanpy源码浅析之pp.calculate_qc_metrics - 何物昂 - 博客园
2、ClusterMap for multi-scale clustering analysis of spatial gene expression