TACOS:一种用于准确预测细胞特异性长的非编码RNA亚细胞定位的新方法
《TACOS:一种用于准确预测细胞特异性长的非编码RNA亚细胞定位的新方法》
《TACOS: a novel approach for accurate prediction of cell-specific long noncoding RNAs subcellular localization》
期刊:《Briefings in Bioinformatics》

摘要
长的非编码RNA(LNCRNA)主要受其细胞定位调节,该细胞定位负责其分子功能,包括细胞周期调节和基因组重排。准确地从序列信息中准确识别LNCRNA的亚细胞位置对于更好地理解其生物学功能和机制至关重要。与传统的实验方法相反,可以将生物信息学或计算方法应用于人类中LNCRNA亚细胞位置的注释。过去,已经开发了几种基于机器学习的方法来识别LNCRNA亚细胞定位,但是鉴定人LNCRNA的细胞特异性定位的相关工作仍然有限。在这项研究中,我们介绍了基于树的堆叠方法炸玉米饼的第一个应用,该方法允许用户识别10种不同细胞类型中人类lncRNA的亚细胞定位。具体而言,我们使用新构建的每种单元格的新构建平衡训练数据集对六个基于树的分类器进行了全面评估。随后,Adaboost基线模型的强度通过堆叠方法集成,并具有适当的基于树的分类器以用于最终预测。与本研究中使用的其他两种方法相比,TACOS在交叉验证和独立评估中表现出一致的性能。可以通过https://balalab-skku.org/tacos访问用户友好的在线TACOS服务器。
Introduction
RNA是分子生物学中央教条中的主要成分之一,在不同的生物学过程中起着至关重要的作用[1]。早期测序数据表明,哺乳动物基因组的> 80%被转录为非编码区域,而仅将较小的部分转录为蛋白质编码RNA [2,3]。由于测序技术和生物信息学分析的进步,已经鉴定出越来越多的非编码RNA(NCRNA),包括圆形RNA,长NCRNA(LNCRNA)和小NCRNA [4-6]。 NCRNA是不具有功能性开放式阅读框架的转录组最大组成部分,但在包括疾病发病机理在内的几种生物学过程中起着有效的作用[1]。基因组经历了广泛的转录,导致成千上万的LNCRNA长度> 200个核苷酸,并且不经过翻译为功能蛋白。它们充当诱饵,增强剂RNA,导向,支架,信号和短肽[7,8]。 LNCRNA可以调节染色质功能,影响信号机制,改变细胞质mRNA的稳定性,并根据核体的定位和与其他生物学的相互作用来调节核体的组装和功能大分子。这些功能中有几种影响了不同生物学和病理生理途径(例如癌症,免疫反应和神经元疾病)的基因表达.
如证明,人类基因组构成> 16 000 lncRNA基因[10];但是,其他消息来源预测> 100 000 lncRNA [11]。迄今为止,只有一小部分的lncrnas被描述了。因此,有必要确定他人的功能表征[9,12]。此外,LNCRNA的多功能功能取决于其亚细胞的局限性。因此,了解LNCRNA的细胞局部局部将有助于破译其潜在的分子机制。
原位杂交(ISH)是一种流行的技术,可以使用标记的互补寡核苷酸探针识别候选LNCRNA的细胞定位[13,14]。单分子f Luorescence ISH是金标准方法,其中使用多个探针来放大f luorescent信号以检测靶RNA,该靶RNA以低水平存在[15]。相反,荧光原位RNA测序方法可在高通量水平上提供原位信息[16]。空间分辨的转录物扩增子读数映射是另一种方法,其中提供了有关完整组织样品中RNA表达的三维位置信息[17]。由于已确定的LNCRNA的数量超过了具有已知本地化的lncRNA,因此有必要实施快速,高效和成本效益的计算方法来协助其识别。
迄今为止,仅开发了几种计算方法来预测组织/细胞系之间的LNCRNA亚细胞定位[18-22]。 Cao等。 [19]提出了一个名为LNClocator的预测指标,该预测因子是根据Rnalocation数据库[23]开发的,以确定五个亚细胞定位。该预测器采用随机森林(RF),支持向量机(SVM)和自动编码器的K-MER频率信息特征。为了构建平衡的训练模型数据集,LNClocator利用了技术synthetic minority oversampling technique[24]。
Synthetic Minority Over-sampling TEchnique(SMOT)解决样本不平衡问题
https://blog.csdn.net/seavan811/article/details/46879783#:~:text=%E4%B8%80%E6%AD%A5%E7%9A%84%E7%A7%AF%E7%B4%AF%E3%80%91-,Synthetic%20Minority%20Over%2Dsampling%20TEchnique,-%E5%8D%81%E5%B9%B4%E7%9A%84%E5%B0%8F%E7%99%BD
2018年,古德纳斯(Gudenas)和王(Wang [20)开发了Deeplncrna,它直接从转录序列预测LNCRNA亚细胞定位。他们通过使用深神经网络提取K-MER频率分析了来自多种细胞类型的链特异性RNA-SEQ样品。 Su等。 [22]通过使用SVM将8支核核苷酸特征纳入一般的Psuedo K-Tupler组成(PSEKNC),开发了ILOC-LNCRNA。最近,艾哈迈德(Ahmad)等人。 [18]通过使用深层局部SVM提取K-MER特征来对四个位置进行分类,从而开发了定位-R。最近,Lin等。开发的LNClocator 2.0 [21]是一种使用可解释的深度学习方法作为细胞系特异性亚细胞定位预测变量。在现有的预测因子中,LNClocator 2.0是唯一可用的细胞特异性预测指标,但它具有足够的改进空间。为了开发基于机器学习(ML)的预测指标,有必要设计适当的编码方法,以表示跨组织/细胞系围绕亚细胞定位的LNCRNA序列片段。
在这项研究中,我们开发了一种基于树的算法,用于细胞特异性长的非编码RNA亚细胞位置(TACOS),以精确检测细胞特异性人LNCRNA亚细胞位置的准确检测,其概述的概述如图1所示。我们为10种不同的细胞类型中的每一种构建了平衡的训练数据集,包括A549,GM12878,H1人类胚胎干细胞系(HESC),HELA.S3(HELA),HEP G2(HEPG),HT1080,HT1080,HUVEC,NHEK,SK,SK,SK,SK,SK,SK,SK .Mel.5(Skmel)和SK.N.SH(SKNS)。利用每种单元类型的平衡训练数据集,我们测试了六个不同的基于树的分类器[RF,极度梯度提升(XGB),ADABOOST(AB),梯度提升(GB),Light GB(LGB)和极为随机的树(ERT)(ERT )]使用10个不同的特征描述符(涵盖组成和理化属性),并确定了基于分类器的基线模型。
随后,我们通过堆叠策略将这10个基线模型与适当的基于树的分类器进行了预测,以进行最终预测。值得注意的是,炸玉米饼是用于识别细胞特异性LNCRNA亚细胞位置的第一个基于树的算法的应用。TACOS将能够协助实验者识别新的LNCRNA位置并阐明其功能更大。

数据集
为了基于序列信息开发预测模型,需要LNCRNA核苷酸序列和定位信息。最近,Lin等。 [21]最近基于核苷酸序列构建了一个高质量的数据集,该数据集具有从Gencode Project [25]获得的可变长度和从LNCATLAS [26]获得的定位信息。为了确定lncRNA的位置,作者使用了不同细胞类型的细胞质/核相对浓度指数(CNRCI),并确定如果CNRCI为> 1,则LNCRNA位于细胞质内,并且如果它为<-1 ,它位于核内。这些数据可通过以下链接获得:https://github.com/yang-j-lin/lnclocator2.对于每种单元格类型,它们通过应用CD-HIT [27] 0.8的阈值来生成非冗余数据集,这意味着这意味着该数据集这些序列没有共享> 80%的序列身份。具体而言,将总数据集分为8/1/1集并用作火车/开发/测试集,其中使用列车和开发设置进行参数优化和模型构建,并使用测试集来评估模型。
我们在当前研究中使用了相同的序列和各自的分类信息,并进行了以下修改:(i)将列车和开发数据集组合在一起,以生成每种单元类型的新训练数据集,从而比阳性样品更大比例。通过利用不平衡的数据集,任何分类器最终都会在交叉验证/培训期间引入类偏见。 (ii)为了避免这种情况,我们考虑了所有正样品和随机选择的等同数量的负样本来自原始样品。 (iii)新培训数据集中的剩余负样本被认为是独立数据集的负样本,并补充了测试集中的相同的阳性样品。表1提供了本研究中使用的数据集的统计摘要,其中培训样本包括相等数量的阳性和负面因素。相反,复制实际情况的独立数据集包含不平衡数据。
特征选择
特征提取是构建ML模型的最关键步骤之一。通常,应在单个数据集上探索多个功能编码[28-31],而不是探索特定编码的收集。在这项研究中,我们研究了每种细胞类型的10种不同编码,并评估了它们将阳性样品与阴性样品区分开的能力。下面提供了每个编码计算的简要说明。
K间距核酸对(CKSNAP)的组成CKSNAP算法计算了由K核酸分离的二核苷酸的频率(K设置为3)。例如,如果k等于0,则会生成16 0个固定的二核苷酸对(‘cu’,‘ca’,‘gc’,‘ga’,ga’,gg’,‘gu’,gu,‘cg’,‘cg’,ug’,‘’ cc’,‘aa’,‘au’,‘ag’,‘ac’,‘ug’,‘uu’,‘ua’)。功能向量定义为:

对于每个描述符,该值对应于给定序列中的二核苷酸。从给定的序列中,二核苷酸MN的频率由RMN表示,并且0间距二核苷酸的总和由S表示。在这里,k在0-3的范围内,间隔为1,生成64D特征向量。
KC是kmer和其他功能的组合。 (i)kmer:kmer编码确定给定序列中存在的可能核苷酸或核苷酸对的数量。先前的研究为kmer计算提供了数学公式[32,33]。设置kmer> 4,可以产生许多功能并遭受维度灾难。我们将kmer = 1(单体),2(二聚体),3(三位体)和4(Teramer)设置,以避免无关紧要和冗余信息。最后,将所有这些KMER组合在一起,导致给定输入序列的340-D(= 4 + 16 + 64 + 256)特征。
(ii)其他功能,包括Z-CURE,GC含量,AUGC比率和GC偏斜,在数学上表示如下:

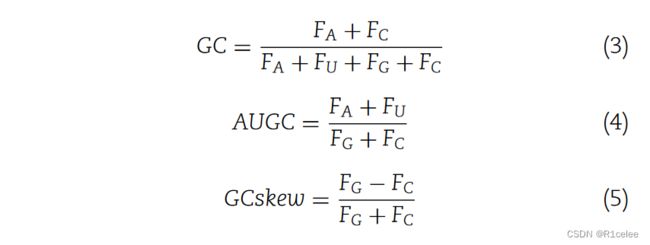
其中FX表示核苷酸X的频率。最后,KC组合了KMER和其他特征以产生346D特征向量。
二核苷酸物理化学特性(DPCP)
使用ILEARN软件包中列出的RNA的21种理化特性(不包括一个自由能之一)[28],我们计算了DPCP:

XM是nth的值(b = 1,2,…,21)RNA二核苷酸物理化学特性(RNA_PCP)。最终,DPCP提供了336D向量。
基于树的ML算法本研究的重点是预测LNCRNA的亚细胞定位,
这是一个二元分类问题。这项研究的目的是确定LNCRNA是位于细胞质还是核中。为了识别最佳ML算法,我们研究了六种基于树的方法:RF [36],ERT [37],XGB,AB,AB,LGB和GB。所有这些分类器已被广泛应用于基于生物信息学序列的函数预测任务[38-40]。重要的是,这些分类器能够比使用归一化特征的SVM和深度学习算法更有效地处理非均衡功能。使用网格搜索和10倍的交叉验证来优化每个分类器的超参数[41,42],该参数搜索范围在补充表S1中提供。实际上,我们重复此过程10次报道了平均性能,并选择了用于构建最终模型的中值参数。这项研究使用Python [43]中的Scikit-Learn软件包来实施RF,ERT,GB和AB。 LGB was implemented in python using the lightgbm package (https://github.com/Microsoft/LightGBM) and XGB in python using the xgboost package (https://xgboost. readthedocs.io/en/stable/python/python_api.html )。
表现评估
有几种广泛使用的性能指标[44-46]可用于测量每个模型的性能,包括准确性(ACC),灵敏度(SN),特异性(SP),Matthew的相关系数(MCC)和接收器下方的区域曲线(AUC)。下面给出了ACC,SN,SP和MCC的数学方程:

TP,TN,FP和FN分别代表了真正的阳性,真正的负面因素,误报和假否定的数量。
结果和讨论评估使用培训数据集对10种不同单元格类型的基于树木的算法评估
E使用10个不同的特征描述符和10倍的交叉验证,对六个分类器(RF,ERT,LGB,GB,XGB和AB)的预测性能进行了全面分析。此外,重复10倍的交叉验证10次报道了10种细胞类型的平均指标(补充图S1 – S10)。根据补充图,S1 – S9,五个不同的编码F4,F5,F6,KC和PSEKNC中的九种不同的细胞类型(HESC除外)具有相似的性能,无论分类器如何,其优于其余编码。然而,无论分类器如何,hESC的八个不同的编码(补充图S10)显示出可比的性能(F1和DPCP除外)。为了获得性能的概述并了解数据集难度的级别,我们为每个分类器提供了10个基于功能描述的模型,结果如图2所示。考虑到我们处理平衡的培训数据集和不平衡的独立数据集,MCC是一个适当的指标,在先前的研究中已建议[47,48]。三种细胞类型(A549,HESC和SKNS)的不同分类器的平均MCC高于0.48,这表明与其余七个细胞类型相比,将细胞质与细胞质区分开相对容易。但是,两种细胞类型(HUVEC和SKMEL细胞)的平均MCC低于0.35,表明它们是对细胞质/核进行分类的最具挑战性细胞类型。此外,平均而言,AB分类器在训练数据集上始终如一地针对10种不同的单元格。
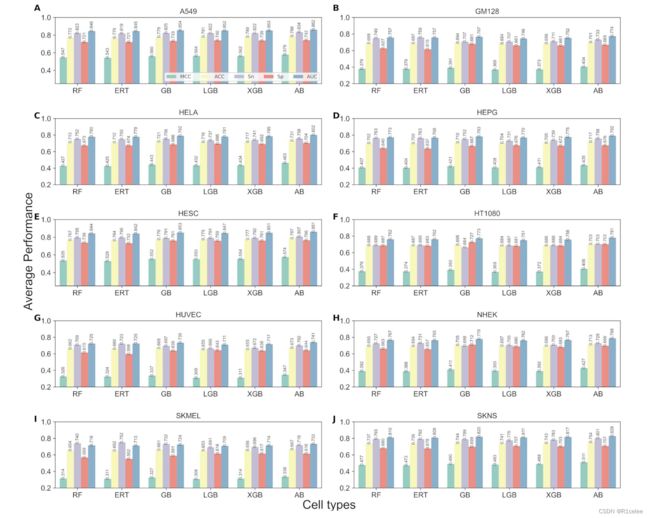
figure2 基于训练数据集的10种不同单元格类型的基于树的算法的平均性能具有10种不同特征描述符。每个A – J列中的每一个都代表不同的单元线,即:A549,GM128,HELA,HEPG,HESC,HT1080,HUVEC,NHEK,SKMEL,SKMEL和SKNS。
为了获得每种单元类型表现最佳模型的更清晰图片,我们为六个分类器中的每个分类器选择了最佳的基于单一FEATURE的模型或基线模型。补充表S2显示了每种单元格中每个分类器的最佳基线模型的性能。显然,AB-PSEKNC模型始终产生最佳的总体指标,包括MCC,ACC和AUC值。 Specifically, it achieved MCC scores of 0.629, 0.455, 0.534, 0.485, 0.619, 0.484, 0.411, 0.492, 0.394 and 0.561, respectively, for A549, GM128, HELA, HEPG, HESC, HT1080, HUVEC, NHEK, SKMEL and SKNS.值得注意的是,其余的大多数分类器也通过PSEKNC编码实现了最佳性能,但指标低于AB,表明在PSEKNC中表现出的歧视模式水平高于其他编码的歧视水平。 。在使用的基于树的算法中,AB分类器有效地从PSEKNC识别了隐藏的模式,从而为所有细胞类型提供了卓越的性能。
使用独立数据集评估每种单元类型的基线模型
我们评估了每种单元格类型的独立数据集上的所有60个模型(6个分类器×10个编码)。图3显示了每个分类器相对于不同细胞类型的平均性能。一般而言,与跨验证性能相比,每个分类器的平均性能下降,而不论细胞类型如何。有趣的是,与所有细胞类型的其他分类器相比,AB分类器的平均MCC始终达到最佳性能,这与交叉验证期间观察到的AB优势一致。值得注意的是,不仅是平均MCC,而且基于AB的最佳单模型都超过了所有细胞类型的同行(补充表S2)。具体而言,单型AB-F4,AB-PSEKNC,AB-PSEKNC,AB-PSEKNC,AB-KC,ABPSEKN,ABPSEKNC,AB-F6,AB-KC和AB-PSEKNC都达到了MCC值0.472,0.277,0.384,0.384,0.320 ,A549,GM128,HELA,HEPG,HESC,HT1080,HUVEC,NHEK,NHEK,SKMEL,SKMEL和SKNS,为0.464、0.316、0.199、0.199、0.271、0.271、0.271、0.271、0.271、0.271、0.271、0.258和0.399。
有趣的是,在独立评估期间,三个编码(F4,F6和KC)能够在四种不同的细胞类型上实现其最佳性能,但是在交叉验证期间,它们的性能相对平均。但是,基于AB的最佳基线模型(AB-PSEKNC)用于六种(GM128,HELA,HEPG,HESC,HUVEC和SKNS),根据交叉验证性能或训练获得了相似的性能水平(以术语为单词在独立评估期间的ACC。这六个物种模型的ACC的绝对差异<6%,表明它们与其他模型相比具有稳健性。通常,选择每种单元格类型的最一致的模型是很简单的。相比之下,我们采用了不同的策略来增强模型的鲁棒性。
探索三种不同策略以改善模型性能
在本节中,我们总结了三种策略,其次是其表演,如下所示:
(i)策略(S1):我们结合了所有特征描述符,并创建了一个混合特征,该特征被馈送到一种基于树的算法中,以使用10倍的交叉验证来开发相应的预测模型。随后,我们比较了六个模型,并选择了最高MCC的模型。在先前的研究[49]中描述的特征选择技术已应用于混合功能,但并未像预期的那样改善预测性能(数据未显示)。因此,将控制(基于混合功能)视为最终模型。
(ii)策略2(S2):基于AB的分类器在交叉验证和独立数据集方面都达到了最佳性能;因此,我们仅根据每种单元格类型的10个特征描述符考虑基于AB的基线模型。集成了从这10个模型得出的细胞质位置的预测概率值,并使用从基于树的分类器得出的适当分类器开发了堆叠模型。
(iii)策略3(S3):与S2不同,考虑了所有六个基于分类器的基线模型。我们总共获得了60个基线模型(6个分类器×10编码),其预测细胞质位置的概率被串联,并将其视为训练六个不同分类器的新型特征矢量,并确定了每种细胞类型的适当元模型。
根据交叉验证和独立评估如表2所示。我们观察到,除了AB外,其他分类器(GB和XGB)在几种细胞类型中的S1表现出色。在S2的情况下,在堆叠方法中至少使用了一次基于树的算法(RF除外)。但是,对于S3,每种细胞类型仅使用三个分类器(RF,GB和ERT)。这些结果表明,在采用各种策略的同时,使用不同的基于树的分类器进行实验很重要。就MCC而言,S3在10个单元格类型的训练数据集上始终优于S2和S1方法(图4A)。

比较来自各种策略的最佳模型的性能。基于(a)中每种类型的单元格和独立评估的三种不同模型[策略1(S1),策略2(S2)和策略3(S3)]进行比较
相比之下,S2的执行始终比独立数据集上的S1和S3方法更好(图4B)。 S2方法比其他两种方法表现出更大的一致性。因此,我们选择了10种细胞类型的基于S2的模型,并将其命名为TACOS。
我们的目标是提高最佳基线模型的性能。因此,我们将它们的表演与炸玉米饼的表现进行了比较。为了获得概述,我们为基线和炸玉米饼模型计算了10种不同单元格类型的平均指标。图5显示,炸玉米饼在交叉验证过程中提高了MCC的1.48%,在独立数据集验证期间将MCC提高了2.61%,这表明堆叠策略改善了两个数据集的总体预测性能。
TACOS与现有方法的比较
我们将炸玉米饼的性能与LNClocator 2.0的性能进行了比较,这是唯一可用的细胞特异性预测指标。重要的是要注意,LNClocator 2.0使用不同的训练数据集进行模型开发,并使用非常小的独立数据集进行了评估,仅报告了AUC值[21]。因此,我们将报道的值与炸玉米饼的值进行了比较。图6显示,在所有10种细胞类型中,炸玉米饼始终优于LNClocator 2.0。 A549,GM128,HELA,HT108,HUVEC,HEPG,NHEK,SKMEL,SKNS和HESC的AUC值的特定改进为1.1、13.5、7.6、10.7、10.6、10.2、6.4、6.4、15.5、9.0和3.4%。根据对于此分析,与现有方法相比,炸玉米饼的改进非常出色
10个概率特征之间的功能相关分析
炸玉米饼模型具有10个不同的特征描述符,每个描述符用于使用AB分类器生成概率特征。接下来,我们评估了10个特征模型是否与不同物种同样相关。图7
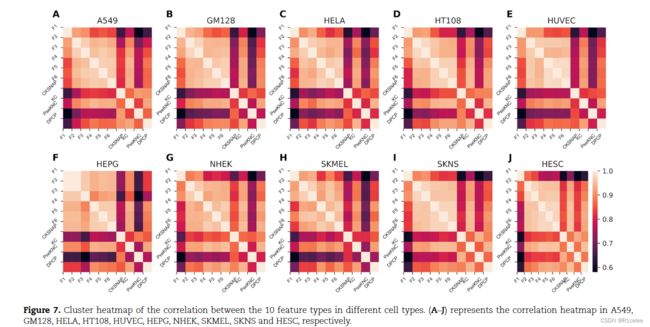
说明了10个概率特征之间相关性的集群热图,该特征为上述问题提供了答案。结果表明,基于F2,F3,F4,F5,F6和KC描述符的概率特征彼此高度相关。然而,基于PSEKNC和CKSNAP的两个概率特征与其他功能中等相关。因此,这些功能相互补充以提高预测准确性。同时,很明显,在所有细胞类型中,特征相关的模式都是相似的。需要基于不同细胞类型的进一步分析,以确认最终预测中不同特征贡献的水平。
功能可视化
我们使用了T分布的随机邻居嵌入(T-SNE)将高维数据转换为两量比图。在训练数据集上,我们将TSNE应用于混合功能以及所有10种细胞类型的概率特征。补充图S11表明,无论细胞类型如何,正面和负样品的杂种特征都是高度重叠的。相反,概率特征(基于AB的基线模型的预测输出)描绘了两个不同的簇,而A549和HESC细胞的两个样品之间几乎没有重叠(补充图S12)。特征分布与性能直接相关,其中A549和HESC的ACC值分别为0.823和0.813。其余细胞类型的ACC在70-80.0%的范围内达到正面和负样本之间的特征重叠仍然存在,这表明采用新颖的特征编码将改善预测性能并区分积极和负偏斜的样本。
跨模型验证
细胞特异性模型通常使用其自己的细胞类型表现出色。具体而言,我们研究了是否可以将细胞特异性模型应用于其他细胞类型。如图8所示,一些细胞特异性模型具有出色的转移性,向其他细胞类型,MCC≥0.50。 GM12878模型在其他六种细胞类型中表现出良好的性能,包括A549,HELA,HT1080,HUVEC,HEPG和SKMEL。同样,HELA模型也可以转移到其他三种类型:HEPG,HT1080和GM12878。此外,A549,HT1080和HUVEC模型可转移到其他两种细胞类型,表明细胞特异性模型在其他类型上的表现也相当出色。但是,四个细胞特异性模型(NHEK,SKMEL,SKNS和HESC)不可转移到其他类型的细胞中,这表明细胞特异性是这些LNCRNA序列的主要特征。基于我们的分析,我们发现高度准确的预测需要细胞特异性模型。此外,这提供了通过组合六种细胞类型(A549,GM12878,HELA,HT108,HEPG和NHEK)来开发通用模型的线索。通用模型也可以通过损害比细胞特异性模型略低的性能来应用于其他细胞类型。
当前研究的局限性
尽管TACOS可以预测细胞特异性的lncRNA定位,但它具有以下局限性:
(i)TACOS是一种基于ML的方法,基于从序列派生的多个手动得出的特征。广泛认识到,ML模型的有效性高度取决于训练过程中使用的特征表示[29-31]。因此,本研究仅考虑组成和理化特性。通过基于其他观点(包括进化信息和新功能),基于广泛的序列分析,可以通过合并功能来实现预测性能的进一步改进。因此,将来,我们计划探索并合并此信息以提高性能。
(ii)TACOS根据给定序列属于lncRNA的假设预测亚细胞定位。在使用炸玉米饼之前,必须使用另一个预测模型从给定的RNA序列中识别LNCRNA。当前,有几种方法可用于识别RNA序列的LNCRNA.[50,51]。建议在使用炸玉米饼之前使用这些方法来识别lncrnas。将来将与炸玉米饼集成一个附加的预测模型,能够根据给定的mRNA序列鉴定LNCRNA和亚细胞定位。
结论
在这项研究中,我们提出了一种称为炸玉米饼的方法,该方法允许使用多个特征编码和基于树的算法准确鉴定人类细胞特异性LNCRNA的亚细胞定位。为了识别最合适的ML算法,我们使用10个不同特征描述符对六种基于树的算法进行了全面的性能评估。平均而言,基于AB的基线模型在交叉验证和独立评估中对所有细胞类型都表现良好。
随后,利用了一种基于树的最佳算法来构建堆叠模型每种单元类型的10个基于AB的基线模型的概率值以提高预测性能。我们的方法与以前的方法不同,因为我们使用平衡的训练数据集开发了炸玉米饼,并使用大型不平衡的独立数据集对其进行了评估。与本研究中采用的其他两种策略相比,炸玉米饼在两个数据集(训练和独立)上的表现始终如一。
炸玉米饼的性能的改善归因于三个因素:(i)探索包括RNA序列信息不同方面的广泛特征描述符,(ii)选择AB特异性基线模型以及(iii)识别适当的分类器用于构建结合基线模型强度的堆叠模型。应该注意的是,采用的基于树的方法在这项研究中,适用于其他基于序列的函数预测问题[52],包括增强子预测[53]和复制起源位点预测[54]。在未来的研究中,如果公开可用的两个以上的数据集大小,将采用与本研究类似的系统方法。此外,我们开发了Tacos Web服务器,并在https:// balalab-skku中免费提供。 org/炸玉米饼。炸玉米饼有望成为实验者识别LNCRNA的亚细胞定位的宝贵工具,这对于进行后续实验以更好地了解其功能将非常有用。