Pan-cancer image-based detection of clinically actionable genetic alternations
目录
摘要
前言
结果
深度学习模型优化
从组织病理图像预测泛癌基因突变
摘要
肿瘤的分子突变可导致肿瘤细胞及其微环境的表型变化。常规组织病理切片可以反映出这种形态学改变。本研究表明深度学习方法能直接从常规病理图像中推断出广泛的基因突变、肿瘤分子亚型、基因表达特征以及病理生物标志物。开发、优化、验证并发布了一站式工作流程,并将其应用于5000名患者的组织病理切片,涵盖多种不同实体瘤。结果表明,针对临床常规的H&E染色病理切片,经过训练的深度学习算法模型能够预测出其中广泛的分子突变。预测结果可以推广到其他人群,同时可以在空间上得以体现。研究将模型部署到移动设备,可为癌症个体化治疗提供即时诊断。更一般地,该方法能够阐明和量化基因型-表型之间的关系。
前言
肿瘤的精准治疗依赖于突变基因检测,临床通过分子生物学检测得到突变结果。但检测周转时间长,所需组织标本及成本消耗大,因此可能成为肿瘤诊疗流程中的瓶颈。临床指南建议对多数实体瘤患者的肿瘤组织进行分子检测。然而,在大多数类型的肿瘤中,常规检测仅包含少数突变基因的检测,比如结直肠癌的KRAS, NRAS和BRAF突变,以及微卫星不稳定。尽管新的研究发现越来越多的临床分子特征,但是目前的临床诊疗流程并未纳入指数级涌现的相关检测方法。例如以往研究已将共识分子亚型(CMSs)确定为结直肠癌的候选生物标志物,但昂贵的测序成本和复杂的检测方法阻碍了其在临床诊疗和临床试验中的广泛应用。因此,肿瘤诊疗越来越需要廉价、可扩展的全新生物标志物。
临床通常难以进行大规模的分子和基因检测,但很容易获取苏木精伊红染色的组织病理切片。本研究认为这些常见的组织切片包含已经明确的和候选的生物标志物信息,而且利用相应的数字化全幻灯片图像(WSIs)可以直接对分子生物标志物信息进行推断。该假设的基本原理是肿瘤细胞的基因突变会引起功能变化,从而影响肿瘤细胞的形态。除了这种一阶基因型-表型相关性之外,肿瘤细胞的基因突变还会影响肿瘤微环境,从而导致更高阶的基因型-表型相关性。这种相关性的一个具体例子为肿瘤微卫星不稳定MSI——临床批准用于结直肠癌免疫治疗的生物标志物。在这个例子中,基因型和表型存在很强的一致性,仅通过观察组织图像中的形态特征就能可靠地推断出相应的基因型。以往关于肺癌、前列腺癌、头颈癌和肝癌等的研究,已经确定了选定基因特征的基因型-表型之间的联系。在此基础上,本研究系统性地研究了所有主要的实体瘤类型,全面探索临床相关分子特征的基因型-表型之间的联系。具体而言,研究探索了能引起足够组织细胞形态反应的分子特征,进而能够通过深度学习算法模型直接从组织病理图像中推断出相应的分子特征。研究的目的是在泛分子,泛癌中使用深度学习,主要关注临床相关的基因分子特征,在较低成本和材料要求条件下,最终得到临床有用的生物标志物。更具体来讲,这种方法能够缩窄要测试的分子范围,增加给定分子特征进入预实验的概率。除了潜在的临床应用,利用组织图像推断基因突变还可以阐明实体瘤中分子改变的下游效应的生物学机制。因此,本研究提出一种深度学习算法模型,直接从组织病理图像中确定分子特征,并对模型进行了外部验证。
结果
深度学习模型优化
研究假设深度学习能从多种不同类型的实体瘤组织图像中直接推断基因突变。为了验证该假设,本研究使用一站式流程训练并验证了深度学习模型(见图1)。为了选择有效的神经网络模型并优化深度学习超参数,结直肠癌微卫星不稳定的预测被用作临床相关的基线任务。在该任务中,使用多种不同的深度学习模型进行超参数调整。出乎意料的是,ShuffleNet——一种轻量级神经网络结构,得到了与复杂网络类似的表现,这些复杂网络包括DenseNet,Inception和ResNet,广泛应用于许多其他研究。ShuffleNet训练时间短,准确率高,针对移动设备进行了优化,可用于分散式即时图像分析,或在显微镜中实时应用。使用426例TCGA结直肠癌患者训练ShuffleNet分类器,并使用379例DACHS队列的患者验证模型,AUROC达到了0.89(95%CI:0.88-0.92),超过了以往研究(AUROC: 0.84),结果表明ShuffleNet是一种有效且强大的神经网络模型,能够直接从组织病理图像中推断临床相关的分子改变。
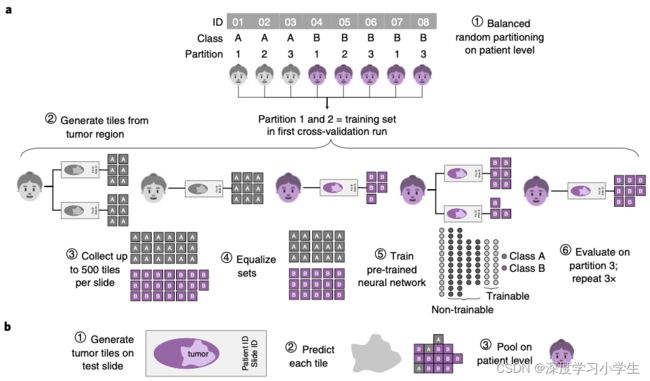 图1.模型流程图
图1.模型流程图
从组织病理图像预测泛癌基因突变
确定深度神经网络超参数之后,系统地将其用于14种不同类型的肿瘤,预测数百种分子突变,在每个队列中进行三折交叉验证。共获得![]() 个独立训练的深度神经网络,用不同类型肿瘤的不同分子特征(基因突变,亚型特征,标准生物标志物)验证这些模型。图2列举了常见的20种突变基因。
个独立训练的深度神经网络,用不同类型肿瘤的不同分子特征(基因突变,亚型特征,标准生物标志物)验证这些模型。图2列举了常见的20种突变基因。
研究首先训练深度神经网络识别这些靶基因中所有的序列变异,结果发现在14种不同类型的肿瘤中,13种肿瘤都至少有一种基因突变可直接从病理图像中得以预测(见图3a-n)。特别是在几种主要的肿瘤类型中,如肺癌、结直肠癌、乳腺癌和胃癌,几种临床和生物学常见的基因突变均可在病理图像中检测到(见图3a-d)。如TP53基因突变(FDR校正![]() ),结直肠癌BRAF基因突变,胃癌候选靶向治疗基因MTOR突变,肺腺癌和胃癌FBXW7基因突变,乳腺癌和胃癌PIK3CA基因突变(小分子抑制剂作为位点,
),结直肠癌BRAF基因突变,胃癌候选靶向治疗基因MTOR突变,肺腺癌和胃癌FBXW7基因突变,乳腺癌和胃癌PIK3CA基因突变(小分子抑制剂作为位点,![]() ),此外在乳腺癌中,从图像中也可以检测到MAP2K4基因突变(MEK抑制剂可能的生物标志物,
),此外在乳腺癌中,从图像中也可以检测到MAP2K4基因突变(MEK抑制剂可能的生物标志物,![]() )。在测试的所有类型肿瘤中,胃癌和结直肠癌可检测到的突变基因数量最多。所有统计学显著的结果中,计算前八种突变结果交叉验证的平均AUROC结果,其中肺腺癌为0.60-0.78,结直肠癌为0.65-0.76,乳腺癌为0.62-0.78,胃癌为0.66-0.78。除此之外,在其他类型的肿瘤中也能检测到一系列显著的突变。模型无法识别出原发性黑色素瘤中的突变基因,反而可以识别出转移性黑色素瘤的FBXW7(
)。在测试的所有类型肿瘤中,胃癌和结直肠癌可检测到的突变基因数量最多。所有统计学显著的结果中,计算前八种突变结果交叉验证的平均AUROC结果,其中肺腺癌为0.60-0.78,结直肠癌为0.65-0.76,乳腺癌为0.62-0.78,胃癌为0.66-0.78。除此之外,在其他类型的肿瘤中也能检测到一系列显著的突变。模型无法识别出原发性黑色素瘤中的突变基因,反而可以识别出转移性黑色素瘤的FBXW7(![]() )和PIK3CA(
)和PIK3CA(![]() )突变。该方法还从前列腺癌组织病理图像中检测出TP53及FOXA1等突变。临床上,KRAS野生型胰腺癌患者是靶向治疗的潜在候选者,该方法能够显著识别出KRAS突变患者,具有很强的临床意义。众所周知,肺鳞癌的分子诊断非常困难,而且几乎没有分子或基因靶向治疗可供选择。因此实验没有得到组织形态学与突变之间的相关性也是合理的,相应地,这种类型的肿瘤中未检测到显著的基因突变。肝癌β-连环基因(CTNNB1)是具有广泛预后和预测意义的关键驱动基因,病理图像可以显著检测到该基因的突变状态。在乳头状肾细胞癌和肾透明细胞癌中,包括KRAS和PBRM在内的多个基因都能被显著检测到,而在chromophobe肾细胞癌中,由于样本量较少,未显著检测到突变基因。在头颈鳞癌中,可显著检测到与细胞死亡抗性相关的CASP8基因突变。在宫颈癌中,可检测到TCERG1,STK11和AMER1等基因突变,并有着较高的AUROC值。
)突变。该方法还从前列腺癌组织病理图像中检测出TP53及FOXA1等突变。临床上,KRAS野生型胰腺癌患者是靶向治疗的潜在候选者,该方法能够显著识别出KRAS突变患者,具有很强的临床意义。众所周知,肺鳞癌的分子诊断非常困难,而且几乎没有分子或基因靶向治疗可供选择。因此实验没有得到组织形态学与突变之间的相关性也是合理的,相应地,这种类型的肿瘤中未检测到显著的基因突变。肝癌β-连环基因(CTNNB1)是具有广泛预后和预测意义的关键驱动基因,病理图像可以显著检测到该基因的突变状态。在乳头状肾细胞癌和肾透明细胞癌中,包括KRAS和PBRM在内的多个基因都能被显著检测到,而在chromophobe肾细胞癌中,由于样本量较少,未显著检测到突变基因。在头颈鳞癌中,可显著检测到与细胞死亡抗性相关的CASP8基因突变。在宫颈癌中,可检测到TCERG1,STK11和AMER1等基因突变,并有着较高的AUROC值。
从组织病理图像中预测泛癌驱动癌基因
因为并非所有的遗传变异都会导致癌症,所以研究再次实验,将突变限制在已经确证的或推定的驱动癌基因。此标准下,出现特定基因突变的病人数量减少,因此满足突变阈值的基因数量也随之减少。相反,因为驱动癌基因和生物学机制密切相关,研究假定其在病理组织形态上有更强的模式。TP53和KRAS等经典癌基因变异绝大部分情况下都属于癌症驱动因子,相应地在驱动癌基因和全面基因突变实验中,这些基因的预测准确性保持类似。除此之外,当局限于癌症驱动基因时,其他基因的突变预测准确性也随之增高。一个典型的例子就是肺腺癌EGFR突变。总之,这些结果说明深度学习可以从多种不同的肿瘤组织病理图像中检测到一系列靶向的和潜在靶向的点突变。
 图2.模型比较(左)和泛癌突变基因预测(右)
图2.模型比较(左)和泛癌突变基因预测(右)
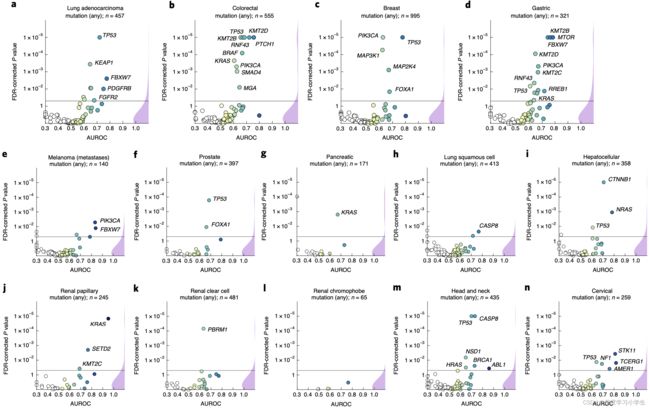 图3.从病理图像直接预测基因突变
图3.从病理图像直接预测基因突变
推断分子亚型和基因表达特征
探究深度学习是否能检测出已确立的肿瘤或免疫细胞分子亚型和基因表达特征。与单基因突变相比,分子亚型和基因表达特征处于更高的功能水平,研究假设它们对于细胞形态的影响比单基因突变更大。为验证这一假设,选取了生物学功能已知和具有潜在临床意义的特征进行研究。例如,CD8阳性淋巴细胞和巨噬细胞等免疫基因表达特征,细胞增殖,干扰素-γ信号传导和转化生长因子-β信号传导。这些生物学过程与免疫治疗等肿瘤治疗反应有关,在组织学图像中检测它们的形态学相关性可以促进设计更为精细的治疗策略。事实上,在研究的所有类型肿瘤中,发现这些高水平的生物学特征比遗传变异或者驱动基因突变的可预测性更强(图4a-d)。
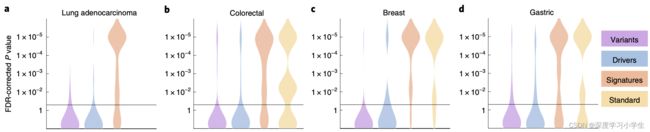 图4 从病理图像推断单基因突变、高水平分子特征
图4 从病理图像推断单基因突变、高水平分子特征
另外,在肺腺癌、结直肠癌,乳腺癌和胃癌中,可预测特征的AUROC值最高(FDR校正P<0.05),见下图e-h。从肺腺癌组织病理图像中能显著检测到细胞增殖特征、巨噬细胞浸润和T细胞浸润特征,并得到较高的AUROC值(下图e)。类似地,结直肠癌、乳腺癌和胃癌组织病理图像中也能显著检测到这些生物标志物(下图f,g,h)。此外,还从胃癌组织病理图像中高度检测出胃干细胞特征(下图h)。近期研究将肿瘤划分成了复杂的分子亚型,本研究方法能够识别肺腺癌的TCGA分子分型,识别AUROC达到了0.74(下图4e);能够识别泛GI亚型,识别AUROC达到了0.76(下图4f);能够识别乳腺癌PAM50亚型,识别AUROC达到了0.78(下图4g)。这些发现可为肿瘤临床试验开辟新的选择,虽然越来越多的证据表明这些肿瘤的分子分型反映了生物学上的不同群体,并与临床结局有关,但是在临床常规治疗和临床试验当中,通常难以获取这些肿瘤细致的分子分型。从组织学检查中检测得到这些亚型,将可直接利用临床试验广泛可用的组织病理图像进行亚型分析,有助于识别对治疗反应的全新生物标志物,或指导特定的分子检测。
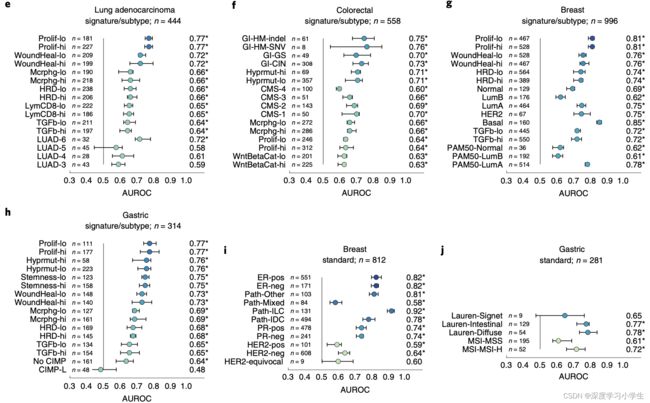 图5 从病理图像推断高水平特征(分子亚型、基因表达特征和标准生物标志物
图5 从病理图像推断高水平特征(分子亚型、基因表达特征和标准生物标志物
使用深度学习预测标准组织病理生物标志物
为了全面评估该深度学习方法临床应用的可行性,研究调查了标准组织病理生物标志物。例如在乳腺癌(图4c,5i)、胃癌(4d,5j)等多种肿瘤中,可以预测得到多数组织病理生物标志物。尤其是乳腺癌组织病理图像可预测激素受体状态(雌激素受体AUROC为0.82;孕激素受体AUROC为0.74)。这些结果说明从常规诊断的组织切片中预测基因改变,高水平分子改变以及已确立的生物标志物是可行的。
其他方法评估
使用深度学习方法从组织病理图像中推断分子特征是一个相对较新的研究领域,通过算法技术改进可以进一步提高预测性能。研究使用结直肠癌队列数据定量评估了其他方法。首先,探究了图像块颜色标准化的作用,和基线方法头对头实验,发现Macenko颜色标准化能够提升模型预测突变的表现,但是亚型和基因表达特征预测效果没有提升;其次,比较了弱监督方法和有标注基线方法的效果,发现弱监督方法仅略低于有标注的方法;最后,分析比较了冰冻切片和诊断切片的预测效果。虽然冰冻切片在临床中并不常见,但是TCGA数据库可提供冰冻切片以供方法比较。在弱监督实验中,发现驱动基因的预测能力是相当的,但从冰冻切片中预测基因突变以及高水平亚型和特征的能力要优于诊断切片。这些数据为将来的大规模验证研究提供了定量指导。
外部验证结果
深度学习方法在单一数据集上容易出现过拟合,部署临床应用之前,需要使用外部数据集验证模型。使用DACHS结直肠癌队列进行外部验证,包括408名患者的HE染色切片、BRAF突变状态和CpG岛甲基化表型(CIMP)。在TCGA结直肠癌数据上训练BRAF以及CIMP深度学习分类器,然后在DACHS数据上验证,验证患者水平的分类准确性。结果表明,仅从HE染色病理图像中就能显著检测到这两种特征,BRAF突变检测AUROC为0.77(0.64-0.82; ![]() ),CIMP-high的分类AUROC为0.66(0.56-0.72;
),CIMP-high的分类AUROC为0.66(0.56-0.72; ![]() ),这说明深度学习预测临床相关的基因特征能够推广到外部人群。
),这说明深度学习预测临床相关的基因特征能够推广到外部人群。
讨论
基于图像的基因检测可作为一种临床和科研工具
研究结果说明,直接从组织图像中基于泛癌深度学习推断广泛的分子和基因特征的可行性。给出的方法在多种临床相关场景中得到可靠的高性能,无需针对特定分子调整技术参数。可以直接从组织图像中推断识别出候选的遗传变异、驱动基因突变、基因表达特征和标准生物标志物,为这些候选生物标志物的大规模验证开辟了前景。作为一项大规模系统性的研究,这项工作识别出了许多与组织图像中可检测的表型显著相关的突变,例如关键致癌通路中TP53、FBXW7、KRAS、BRAF和CTNNB1的产物。除了能识别出单独的基因突变,还能从组织图像中推断出高水平的基因表达亚型或者特征。其中许多亚群都代表了已经确立的具有不同生物学特征的患者,比如结直肠癌的CMSs分子亚型。通过将这些分子定义的亚组与特定的组织图像特征联系起来,我们的方法构建了一个工具,可以破译实体瘤中分子改变引起的下游生物学效应。TCGA图像数据训练得到的模型能够外推到外部数据,说明给出的方法有望应用于真实世界队列的常规检查标本。值得注意的是,在临床工作流程中实施基于组织图像的深度学习方法,还需要额外的回顾性和前瞻性验证,以及监管机构的批准。例如,临床可将该方法作为预筛选工具,富集特定分子测试的患者群体。虽然深度学习技术在临床的首次应用将涉及肿瘤组织或者感兴趣区域的识别,但是我们的方法能够很容易的添加到这种数字化病理流程中,为组织病理数字化诊断提供额外的动力。
缺陷
本研究方法目前的一个缺点是,对于一些分子特征的预测,AUROC值较低。提高预测准确性的一个策略是在更大的患者队列上再训练模型。再训练有望提升模型性能,因为以往研究表明深度学习模型在组织病理图像中的性能与训练队列中的患者数量成正比。此外,算法技术改进也能改善深度学习模型的性能。前面对其他方法的评估为此提供了多个层面的指导,首先是关于神经网络模型的选择,结果表明轻量级的神经网络模型性能与复杂模型相当,有助于在分散式设备上进一步评估这些方法。虽然这一结果基于临床相关的基准任务并推广到外部人群,但是我们无法排除其他性能更好的模型出现。其次是关于输入图像数据的类型,以往研究使用了冰冻切片,我们的方法则使用了福尔马林固定石蜡包埋的组织切片(对应TCGA数据库中的诊断切片),因为这种切片在临床更为常见。临床中,冰冻标本仅占病理标本的一小部分,因此针对石蜡包埋针对切片的方法对于大规模的临床验证至关重要。头对头比较的结果表明使用冰冻切片作为输入图像通常结果更好,这是基于石蜡包埋方法的一个缺陷,需要更多的研究来探究这一结果的原因。最后,研究的基线方法依赖专家标注的肿瘤组织,模型的应用受到限制,只能从侵袭性肿瘤组织中进行学习。这种设计背后的基本原理是,尽管计算机视觉取得了进步,但是肿瘤组织的专家标注仍然是组织病理研究的金标准。但是,头对头研究结果表明无须专家标注的弱监督学习方法并没有显著降低模型的性能,这说明了更加简单的数据处理流程是可行的。最后,在可扩展性和可重复性方面,全自动的工作流程有望优于手动工作流程,我们公开了方法的源码,可供在更大规模数据上进一步优化和验证。
破译基因型-表型关系
除了可作为临床应用工具,深度学习方法从组织病理图像推断分子特征还可以揭示肿瘤生物学的更多基本特征。研究系统筛选了数百种分子改变,并确定了与组织图像中可检测特征模式相关的候选分子改变,利用预测热力图(图6a-e)可以观察到这些特征模式,这种将基因预测空间化的方式是传统bulk基因检测所缺乏的一个关键方面,可用于追踪特定空间区域内的分子改变。还可以通过另外一种方法对模型预测结果进行解释,即可视化排名靠前的图像块(图7),这种方法有助于发现新的形态学特征。实际上,结直肠癌CMS亚型的图像块中,预测为CMS1的图像块中包含低分化肿瘤组织,预测为CMS2-3的图像块中包含高分化腺体,预测为CMS4的图像块中包含高级别基质成分,这些模式对应CMS亚型已知的生物学过程,证实了深度学习模型能够检测到具有生物学意义的特征。类似地,对BRAF突变患者排名靠前的图像块进行了可视化,其中显示了低分化区域以及粘液区域,这与以往的研究一致。
对胃癌中排名靠前的图像块进行可视化,显示细胞高密度区域与增殖基因表达特征相关,同时还识别出与突变有关的特征模式,例如(AMER1和MTOR突变的特征模式),有利于理解特定基因如何影响细胞行为和形态。此外,14种不同类型肿瘤的预测性能存在显著差异,样本量大小的差异是其中一个原因,但额外的生物学效应的差异也可能导致性能差异,一个假设就是具有很少临床靶向突变的肿瘤(如肺鳞癌和胰腺癌)也显示出很少的可检测突变,有必要进一步对其进行研究。
结论
综上,研究证实了仅使用深度学习就能从常规的组织病理图像中推断出实体瘤的分子改变。这可能成为阐明肿瘤基因型-表型关系的有用工具,并最终为临床试验和临床常规工作流程提供低成本的生物标志物。