实用生物信息学2:多组学数据整合和挖掘
实用生物信息学2:多组学数据整合和挖掘
本文部分/全部图片与文字来源网络或学术论文,仅供学习使用,持续修改完善中。
文档下载见:生信-国科大雁栖湖春季课程《使用生物信息学2:多组学数据整合和挖掘》知识点整理-其它文档类资源-CSDN文库
目录
实用生物信息学2:多组学数据整合和挖掘
第一章生物数据的存储与可视化
(1)生物信息学的定义及科学地位
(2)HTML(超文本标记语言HyperTextMarkupLanguage)
(3)Linux
(4)PHP编程
(5)E-R型数据库
(6)构建简单网站的基本操作步骤
(7)R语言
基本数据类型:
数据结构:
Tidyverse数据科学工作流程:
R语言优点:
(8)ggplot数据可视化
可视化编码:
色彩空间:
ggplot的图形语法:
ggplot2图形语法特点:
(9)管道运算符(%>%)
(10)Bioconductor软件包
利用BioC进行高通量基因组学数据分析的几个方向:
(11)python
数据类型:
常见的包:
第二章序列分析
(1)序列比对
两个序列的比对流程:
1、暴力法:
2、点阵法:
3.动态规划法:
空位罚分
打分矩阵
4、FASTA算法——数据库比对
5、BLAST算法——局部序列比对
Blast搜索种类:
6、BWT算法——短序列比对
(3)数据库序列搜索的基本思路
(4)Motif基本概念
(5)模件搜索的方法
(6)描述Motif的几种常用方法
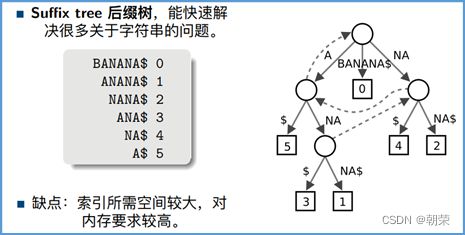
(7)序列库的主要索引方法
(8)NGS序列分析软件
(9)RNA-seq数据分析步骤
(10)序列富集分析技术
(11)DNA调控元件的主要类型
(12)鉴定调控元件的常用实验方法
DNA序列分析:
生物化学注释
eQTLmapping
(13)转录因子和转录因子结合位点的识别
(14)调控元件相关实验方法
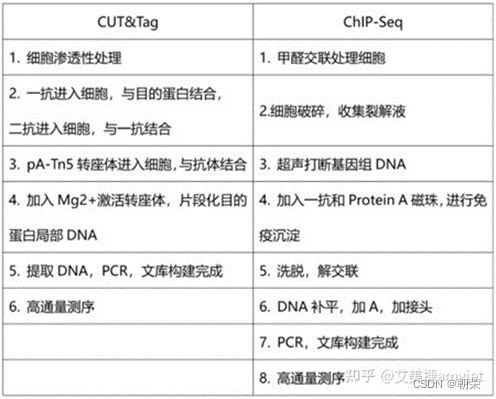
1、Chip-seq:
2、CUT&Tag:
3、ATAC-seq染色质可及性
(14)序列关联分析:
染色质构象的基本概念
染色质成分:
染色质结构:
染色质分类:
主要相关实验方法
3C-Seq:
Hi-C:
第三章多组学数据的整合分析
(1)转录组数据分析:
数据整合的基本思路:
(2)微阵列
(3)BeadChip
(4)归一化
1.Scaling:
2.Non-linearmethod非线性方法:
3.QuantileNormalization算法实现
三种归一化方法的评估:
RNA-seq中的归一化:
(5)基因差异表达分析
差异表达分析的统计学原理
t检验
差异表达分析方法
1.SAM:微阵列的重要分析。
2.RP:RankProducts。
芯片技术固有的缺陷包括:
RNA-seq面临的挑战
(6)NewTuxedo套件差异表达分析
(7)聚类分析和主成份分析
层次聚类的基本思想和算法实现
分割聚类的基本思想和算法
PCA的基本思想
PCA步骤:
(8)单细胞转录组测序分析
实验的基本步骤
scRNA-seq的缺陷:
scRNA-seq分析的基本步骤:
单细胞测序数据分析:
UMI的原理(重要的地方)
质控
单细胞测序数据批次效应处理
(9)单细胞表观组学及多组学整合分析
单细胞表观组主要技术
空间转录组主要编码方式
(10)基因调控网络的基本概念
基因调控网络组成:
模型的几种主要形式
GRN基因调控网络解析
第四章多组学数据的深度分析
(1)机器学习的计算原理
监督学习(当训练样本带有标签时):
非监督学习(训练样本全部无标签时):
(2)感知机的关键特征
单层感知机:
多层感知机:
激活函数
(3)卷积神经网络与图像识别:
反向传播算法
算法原理:
卷积运算的基本概念
(4)生物影像组数据的深度处理:
卷积神经网络的常用框架
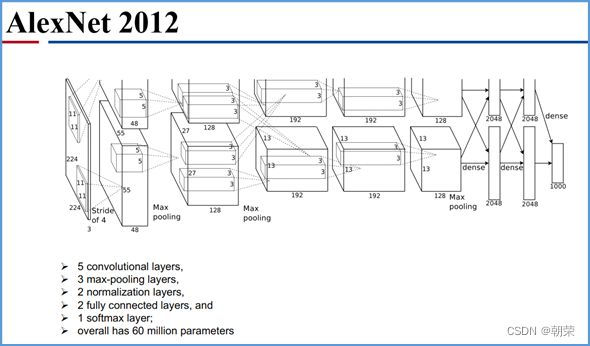
卷积神经网络AlexNet的关键特征
AlexNet特点:
Dropout
(5)神经网络的应用与局限性:
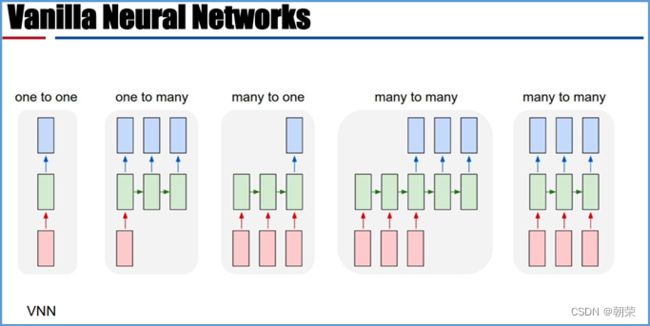
序列神经网络的特点
RNN
LSTM长短时记忆神经网络
第一章生物数据的存储与可视化
1.计算技术与操作基础:生物信息学定义及学科地位,XAMPP(Apache+MySQL+PHP+PERL),Linux/Web服务器/HTML/PHP/ER数据库基本概念,ER数据库的键/主键/外键,构建简单网站的基本操作步骤
2.R语言基础:R/Bioconductor基本概念
3.R语言数据可视化基本原则:ggplot图形语法含义,常用图表类型
4.Python基础:Python基本概念,常用库(关注重点讲的内容)
(1)生物信息学的定义及科学地位
定义:生物信息学是以生物信息的存储、展示、分析、以及深度挖掘为主要任务,利用网络服务器、数据库、编程等多种计算机技术来完成这些任务的学科。
地位:是一门根植于现代生物学、统计学、以及计算机科学的交叉学科。从其诞生之日起,它就具有与生物物理学和生物化学同等重要的地位。
(2)HTML(超文本标记语言HyperTextMarkupLanguage)
- HTML不是一种编程语言,而是一种标记语言。
- 一个HTML文件就是一个网页。
- HTML文件包含两部分:html标签和文本text。
- Web浏览器的作用是读取HTML文档,并以网页的形式显示出它们。浏览器不会显示HTML标签,而是使用标签来解释页面的内容。
- HTML可互作性:通过表单(form)用户可以和后台程序、数据库发生相互作用,可以实现网页的动态展示。
- 平台不依赖性:独立于计算机硬件和操作系统。
- 可读性:丰富的标题、字体、颜色、图片、布局等的格式。
- HTML的表现由尖括号包围的关键词,标签通常是成对出现的。
与之间的文本描述网页
与之间的文本是可见的页面内容
与之间的文本被显示为标题
与
之间的文本被显示为段落(3)Linux
常用的命令:
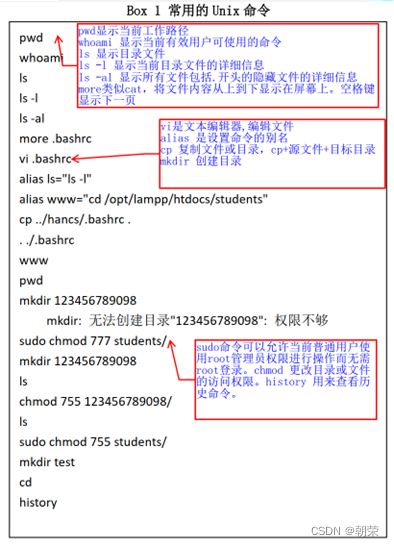
(4)PHP编程
编程语言的发展史:机器语言(0/1代码),汇编语言(将0/1代码编译为机器代码),高级语言(即编程语言,C,C++,JAVA等,更加封装为程序员可编写的),高级语言(与人类语法相似,PHP,PYTHON,C#等)。
PHP的定义:
- PHP是"PHPHypertextPreprocessor"的首字母缩略词;
- PHP是一种被广泛使用的开源脚本语言,尤其适用于Web开发;
- 它的语法利用了C、Java和Perl,易于学习;
- Client-Server模型中,PHP脚本在服务器端而非用户端运行;运行的结果以纯HTML文本返回浏览器
PHP的功能:
- PHP文件能够包含文本、HTML、CSS以及PHP代码。
- 能生成动态页面内容。
- 能够对服务器上的文件进行打开和关闭,还有增删查改操作。
- 能够接收表单(form)数据。
- 能够发送并取回cookies;
- 能够对数据库中的数据进行增删查改;
- 能够限制用户访问网站中的某些页面;
- 能够对数据进行加密;
- PHP可以不受限于只输出HTML。能够输出图像、PDF文件、甚至Flash影片。
(5)E-R型数据库
数据:对物体(object)或事件(phenomenon)某种属性(attribute)的定性描述或定量测量结果。
数据库:是用来组织、存储、查询数据的软件系统。数据库的理论基础是关系代数。数据库不同于一般文件系统的最大特点就是它是基于模型(某种数学模型)的数据存储方式。
E-R型数据库:是以事物和彼此之间关系为模型的数据库。该模型认为任何事物都具有一定属性并且和其他事物发生某种联系。E-R数据库的基本单元是表,表可以代表一类entity,或者一类entity的一种复合属性,或者两类entity之间的关系。

数据库管理系统DBMS:是管理数据库的软件系统,它对数据库的使用、安全、备份等提供各种操作手段。
数据库设计的过程:
| 1.收集用户需求,整理出所有的概念。 |
| 2.鉴定出哪些概念是实体,哪些是其属性,哪些又代表了实体之间的关系。把这些概念转化成相应的数据库表。 |
| 3.确立键(key),主键(primarykey,PK)和外键(foreignkey,FK)
|
| 4.明确一对多(1:N)和多对多(N:M)的关系。 |
| 5.数据库规范化。 需满足数据库三范式(防止数据冗余,数据冲突):
|
数据库语句sql:
数据定义语言(DataDefinitionLanguage,DDL)
CREATEDATABASEseqdb
CREATETABLEprotein(
idINTPRIMARYKEYAUTOINCREMENT
seqTEXT
lenINT)
ALTERTABLE...
DROPTABLEprotein
DROPDATABASEseqdb
数据操作语言(DataManipulationLanguage,DML)
SELECT:查询
UPDATE:修改更新
INSERT:插入,增加
DELETE:删除
(6)构建简单网站的基本操作步骤
1.构建网络服务器webserver,能够在后端处理用户信息,发送给用户所需求的信息。作业中是放在opt/lamp/htdoc/s123456目录下,这是老师提供的服务器。也可以在本机电脑上安装phpstudy软件,大考apache和mysql服务。
2.编写HTML静态页面代码文档,通过表单from链接到php代码,可以使用notepad++软件。
3.编写php代码文档,实现数据库访问和操作,将得到结果反馈到页面。使用notepad++软件。
4.将编写好的所有代码部署到服务器目录下。
(7)R语言
R是一个用于数据分析和做图的软件系统。R是一个解释型高级语言,包括了分支、循环、函数等高级语言的功能。R可以调用其他语言写的程序。R包含大量的统计学方法。R具有包含做图在内的多种数据展示方法。
基本数据类型:
字符型character:'a',"good","TRUE",'23.4',引号来定义
整型integer:存储正数,在数字后面加大写字母L申明以整型方式储存。
双整型double:储存普通数值型数据,可正可负,可大可小,可含小数可不含
数值型numeric:12.3,5,999
复数型complex:复数类型,即形如:1+i类的数据
逻辑型logical:TRUE,FALSE(区分大小写)
原始型raw:用来存储数据的原始字节
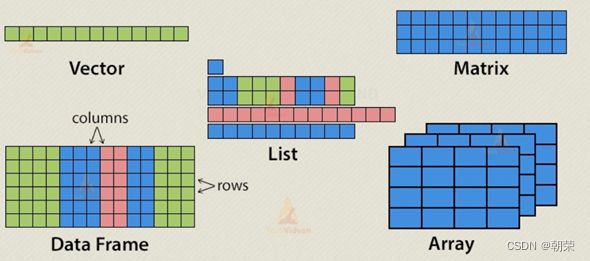
数据结构:
- 向量vector:c()创造向量的函数。访问下表从1开始,a[1:4]是取出第1-4项,包含第1和第4项
- 矩阵matrix:一张表格。
- 数组array:虽然矩阵被限制为二维,但阵列可以具有任何数量的维度。数组函数使用一个dim属性创建所需的维数。
- 数据框dataframe:数据帧是表格数据对象。与数据帧中的矩阵不同,每列可以包含不同的数据模式。第一列可以是数字,而第二列可以是字符,第三列可以是逻辑的。它是等长度的向量的列表。使用data.frame()函数创建数据帧。
- 列表list:列表是一个R对象,它可以在其中包含许多不同类型的元素,如向量,函数甚至其中的另一个列表。
- 因子factor:因子是使用向量创建的r对象。它将向量与向量中元素的不同值一起存储为标签。标签总是字符,不管它在输入向量中是数字还是字符或布尔等。它们在统计建模中非常有用。使用factor()函数创建因子。nlevels函数给出级别计数。
Tidyverse数据科学工作流程:
1.导入(import)
2.整理(tidy):按照一定的格式存储数据。
3.转化(transform):提取特定的观察,创建新的变量,计算一些统计量。
4.可视化(visualization):对原始数据做一些基本作图,看到数据的一些基本特征。
5.建模(models):机器活动。局限性:任何模型都是有假设前提的!
6.展示/交流(communication):把你的分析结果展示给别人。
7.编程(programming):编程是数据分析每一步不可缺少的。

R语言优点:
- 免费、开源。
- 语言结构相对松散,简单易学。
- 强大的作图能力。
- 社区强大,非常丰富的软件包,例如Tidyverse,Bioconductor。
- 优秀的集成集成开发环境RStudio。
(8)ggplot数据可视化
可视化:将数据以一定的变换和视觉编码原则映射为可视化视图。
可视化编码:
是可视化的核心内容,是将数据信息映射成可视化元素的技术,其通常具有表达直观、易于理解和记忆等特性。由两部分组成:标记和视觉通道。
- 标记:代表数据属性的分类,通常是一些几何图形元素,例如:点、线、面、体。
- 视觉通道:人眼所能看到的各种元素的属性,用来展示属性的定量信息,通常有位置、长度、角度、方向、面积、体积、饱和度、色相、纹理、形状。


色彩空间:
是描述使用一组值(通常3-4个值)表示颜色的方法的抽象数学模型。
- RGB色彩空间:采用笛卡尔坐标系定义颜色,三个轴分别对应红色(R)、绿色(G)和蓝色(B)三个分量。
- CMYK色彩空间:青色(Cyan)、品红色(Magenta)、黄色(Yellow)和黑色(Black)。
- HSV色彩空间:色相Hue,饱和度Saturation,明度Value
- HSL色彩空间:色相Hue,饱和度Saturation,亮度Lightness
五种基本作图:
散点图:又名双变量图,揭示两个数值变量的关系。
折线图:自变量(比如时间)不同且有顺序。
直方图:
箱线图:
条形图:

ggplot的图形语法:
数据部分:
- 数据:输入的数据。
- 集合对象:表示数据的几何图形。点、线等。
- 视觉通道:几何的视觉特征。尺寸、颜色、形状等。
- 度量:视觉特征转换为显示值,坐标轴度量和图表原色度量。
- 统计:对数据进行聚合和其他极端。总数,均值等。
- 坐标:确定几何位置的数值系统。直角坐标系、极坐标系,地理坐标系。
- 分面:将数据拆分为子集。
美化部分:
Theme调整图表主题。
Guide调整图例。
添加标签,自注释,备注等。
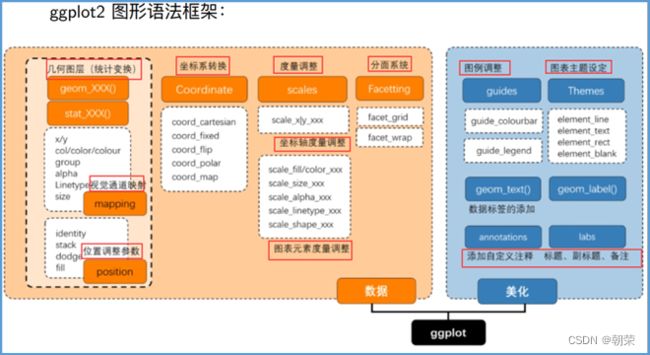
ggplot2图形语法特点:
- 采用图层的设计方式,有利于结构化思维实现数据可视化;
- 将表征数据和图形细节分开,能快速将图形表现出来;
- 图形美观,扩展包丰富,易于定制个性化图表。
(9)管道运算符(%>%)
x左侧的变量%>%将作为右侧函数中的第一个参数应用。可以使用点(.)称为占位符的默认行为进行更改。
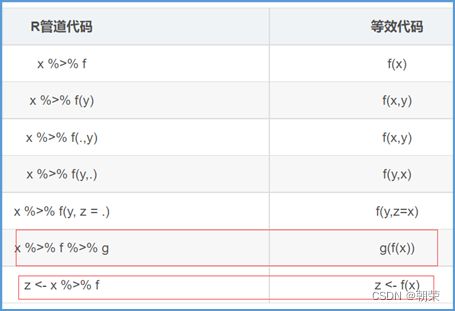
x%>%f(y)turnsintof(x,y),
x%>%f(y)%>%g(z)turnsintog(f(x,y),z)
(10)Bioconductor软件包
是一个基于R系统的用于进行高通量组学数据分析的平台。
安装方法:
install.packages("BiocManager")
BiocManager::install()
BiocManager::install("GenomicRanges")
优点:以R语言为基础,高质量的文档,丰富的统计和作图方法,方便的注释系统,多种培训课程,开源项目,开放式的开发系统。
利用BioC进行高通量基因组学数据分析的几个方向:
- 高通量测序数据分析。
- 多样品的协同分析。
- 注释数据的软件包和资源。
- 实验数据包。
- 各种组学数据整合分析。
- 可视化。
- 可重复研究。
- 替代和补充工具。
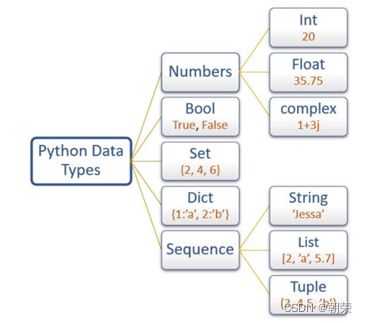
(11)python
数据类型:
不可变数据(3个):Number(数字)、String(字符串)、Tuple(元组);
可变数据(3个):List(列表)、Dictionary(字典)、Set(集合)。

常见的包:
- Numpy:可用来存储和处理大型矩阵。
- Pandas:是基于NumPy的一个数据分析包,包含大量库和一些标准数据模型,提供了高效地操作大型数据集所需的工具。
- Matplotlib:是一个Python的绘图库,数据可视化基础。可以生成绘图,直方图,功率谱,条形图,错误图,散点图等。
- Seaborn:基于matplotlib的数据集分布可视化库,可以绘制进阶图形。
- Scipy:是一个开源的Python算法库和数学工具包。包含的模块有最优化、线性代数、积分、插值、特殊函数、快速傅里叶变换、信号处理和图像处理、常微分方程求解和其他科学与工程中常用的计算。
第二章序列分析
1.序列比对分析:序列比对基本概念,双序列比对的几种基本算法(全局的,局部的),动态规划的核心思想/要点/手工实现(会计算),数据库序列搜索的基本思路。(怎么提高速度)
2.序列模式分析:Motif基本概念,描述Motif的几种常用方法
3.转录本剪切模式分析(NGS序列分析):序列库索引的几种基本算法,BTW算法手工实现(excel矩阵变换,移位),NGS序列分析的几种常用软件(屠老师的看PPT的小结,题目所在)
4.序列富集分析(调控元件):DNA调控元件的主要类型,鉴定调控元件的常用实验方法
5.序列关联分析:染色质构象的基本概念,主要相关实验方法
(1)序列比对
序列:一个序列就是按照特定顺序排列的一组事物。
序列比对:将两个或多个序列排列在一起,标明其相似之处。序列中可以插入间隔(通常用短横线“-”表示)。对应的相同或相似的符号(在核酸中是A/T/U/C/G,在蛋白质中是氨基酸残基的单字母表示)排列在同一列上。
两个序列的比对流程:
- 对于两个长度为m和n的序列S1和S2。
- 建立S1和S2中所有元素的某种对应关系,允许一个元素和一个空格(gap)对应,但是不允许来自两个序列两个gap对应。
- 形成新的序列。在这种对应关系确立后,由于gap的出现,S1和S2分别转变成了S1’和S2’;
- 建立某种打分规则,如相同的元素对应得2分,不同的元素对应得1分,元素和gap的对应得0分;
- 计算当前两个序列比对结果的总分。
- 采用不同的比对选择,获得所有S1和S2的对应关系的总分。
- 选择最大的总分,相应的对应关系就是这两个序列的比对结果。

(2)双序列比对的方法

1、暴力法:
实际上是穷举法,就是说把定义中提到所有的对应关系找到,然后找出得分最高的对应关系。
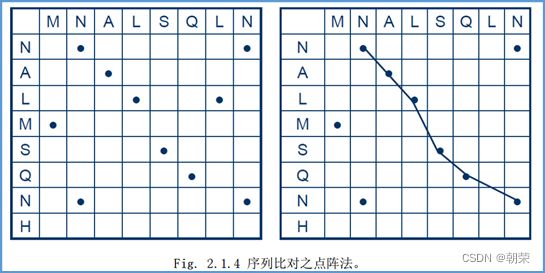
2、点阵法:
对于两个长度为m和n的序列S1和S2。构建一个m*n表格,行列元素相同则打点,选取点数最多的那条折线就是两个序列的比对。
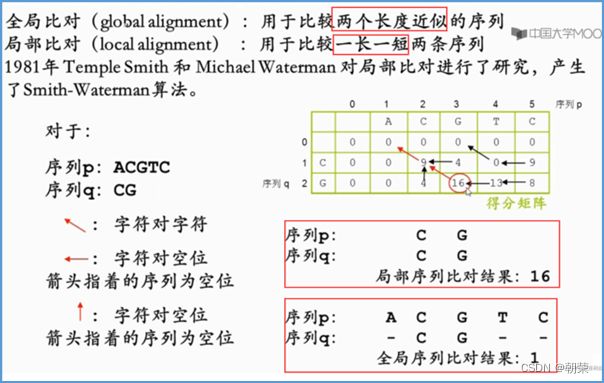
3.动态规划法:
全局比对的动态规划算法(Needleman-Wunsch算法):对两条序列的全长都进行比对

局部比对的动态规划算法(Smith-Waterman算法):获得匹配最好的局部序列片段。

空位罚分
当gap开头罚分小,gap延长罚分大的时候,做出来的比对里面gap很分散,极少有连续长串的gap出现。
当gap开头罚分大,gap延长罚分小的时候,说明在连续的字母里插入一个gap打开一个缺口要付出很大的代价,因为gap开头罚分大。gap都集中连成长串出现。
所以,可以通过调整gap开头和gap延长的罚分,可以把序列比对做成我们期待的样子。
打分矩阵
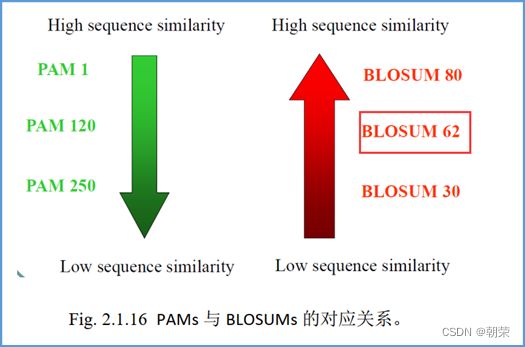
PAM:基础的PAM-1矩阵反应的是进化产生的每一百个氨基酸平均发生一个突变的量值,由统计方法得到。
如果序列亲缘关系远,序列间会有很多突变,PAM后面跟一个大数字的矩阵。
BLOSUM:BLOSUM矩阵都是通过对大量符合特定要求的序列计算而来的。BLOSUM矩阵的相似性是根据真实数据产生的。在BLOSUM80中的80,代表这个矩阵是由一致度≥80%的序列计算而来的。同理,BLOSUM62是指这个矩阵是由一致度≥62%的序列计算而来的。
亲缘关系远的序列,BLOSUM后面跟一个小数字的矩阵,相似度低。
亲缘关系较近的序列之间的比较,用PAM数小的矩阵或BLOSUM数大的矩阵。
亲缘关系较远的序列之间的比较,用PAM数大的矩阵或BLOSUM数小的矩阵。
如果关于要比较的序列你不知道亲缘关系远近,那么就闭着眼睛用BLOSUM62。

4、FASTA算法——数据库比对
查询序列与一个数据库中的大量序列进行比对的结果。
思想:
1、找出所有的长度为K的完全匹配的短序列。
2、利用PAM250等为这些短序列打分并找到在对角线方向的得分从高到低排列的前十个。
3、把一些短序列合并成带有GAP的长比对。利用最长路径算法找到图中长度最大的路径。
4、在长度最大的路径两侧设定一个区域,利用DP获得的分比长度最大的路径还要高的比对作为最终的比对结果。

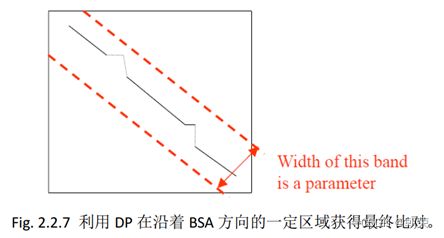
5、BLAST算法——局部序列比对
BLAST快速的数据库相似性搜索工具。它可以在尽可能准确的前提下,快速的从数据库中找到跟某一条序列相似的序列。
思想:是从一些小的高度匹配的区段(highscoringwordpair,HSW)向外扩增以获得最佳比对。
- 查询序列的预处理。先规定HSW的长度,比如3(W=3)。找出查询序列的上所有3-words,对每个3-words找出与它相似的words(Neighborhoodwords,NBWs),建立所有相似性不低于某个阈值的3-words与其NBWs的查询表。
- 扫描数据库序列。对于数据库序列包含的3-words通过查询序列的查询表找到HSWPs。
- 获得最佳比对。将一个HSWP向两边扩展,直到得分低于某个预设值或者E-value,这个扩展的比对叫做HighScoringSegmentPair最高部分匹配片段。将HSWP扩展为HSP。


Blast搜索种类:
BLASTN:qS和dSs皆为DNA序列。
BLASTP:qS和dSs皆为蛋白质序列。
BLASTX:qS为DNA序列,dSs为蛋白质序列,DNA要翻译成6种蛋白质序列后进行比对。
TBLASTN:qS为蛋白质序列,dSs为DNA序列,DNA要翻译成6种蛋白质序列后进行比对。
TBLASTX:qS和dSs皆为DNA序列,qS和dSs的DNA都要翻译成6种蛋白质序列后进行比对。

6、BWT算法——短序列比对
BWT算法可以分为编码和解码两部分。
编码:
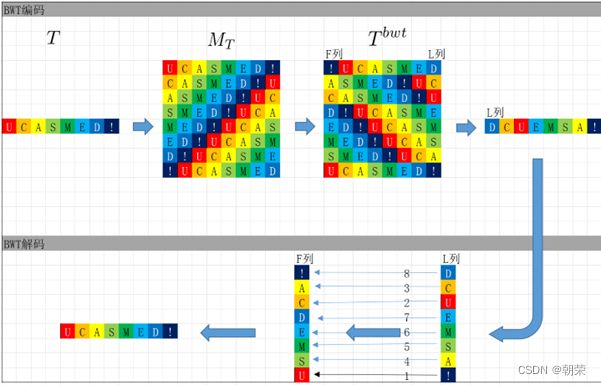
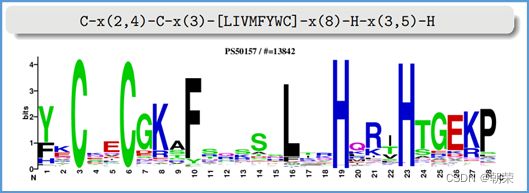
DNA的字符集是(G,A,T,C),给T的末尾加上一个特殊字符“$”以表示T的结尾。字符集就是(G,A,T,C,$),字符可以按照某种规则来排定它们的大小顺序。比如规定$ 思想: 1、T=abaaba$,把T向左旋转一个字符得到了一个新的序列T1=baaba$a,再向左旋转一个字符得到T2=aaba$ab,重复得到T3,T4,T5,T6,T7=T 2、把T和T1,2,3,4,5,6从低到高摞起来形成一个m*m的矩阵。按照前面规定的字符集的大小(从小到大)对矩阵的行进行排序。新的矩阵叫做BWM,其最后一列就是BWT(T)=abba$aa。 解码: 只需要转换后的BWM矩阵的首列F列和尾列L列,从L列的$开始找列中对应的值放在第一位,以此列推,直到找到F列中的$结束,就能还原出原来的T序列。 思想:利用短片段匹配(word)进行过滤和快速定位,再延伸,从而大幅提高比对速度。 模件Motif:是一段特征序列,例如转录因子TF及其在DNA上的结合位点TFBS,还有真核生物中经常有多个TFBS彼此相邻形成顺式调控元件CRM,TFBS和CRM这类具有某种特征的序列就成为模件。 1)从头搜索法。不需要任何先验知识,直接从兴趣集和背景集出发来搜寻任何新的和已知的模件。有助于发现新的模件,但是计算量大,即使发现新的,也不知道对应的转录因子是什么。 2)已知模件的富集分析。前提是有一个已知模件数据库,我们要看看哪些已知模件相对于背景集在兴趣集中有所富集。这种方法计算量小,不能发现新的模件。 具体的算法有: MEME套件:使用EM算法,以一组DNA或蛋白质序列(训练集)作为输入,输出发现的Motif。 (1)一致序列:多序列比对结果中每一列出现最多的碱基或者氨基酸(或IUPAC兼并码)构成的序列,是一条单一序列;是motif的最简单描述。 (2)正则表达式:来描述Motif。正则表达式使用单个字符串来描述、匹配一系列匹配某个句法规则的字符串。 (3)位置频率矩阵PFM:模件上每一个碱基位置上不同碱基出现的频率不同,可以用位置频率矩阵PositionFrequencyMatrix(PFM)来表示motif。 (4)位置概率矩阵PPM:在位置频率矩阵的基础上,将每个频率换算成0-1之间的概率,计算每个碱基的概率得到位置概率举证PPM。 (5)位置权重矩阵PWM:上面PFM和PPM方法没有反映出模件出现的概率与背景序列的不同,令Mk,j=log2(Mk,j/bk),bk是背景序列,由该数值构成的矩阵叫做位置权重矩阵(positionweightmatrix,(PWM) (6)使用序列logo表示motif。 基于哈希表的索引Hash-based(Salmon,Kallisto):哈希表是根据键(Key)而直接访问在内 存存储位置的数据结构。能够快速查找序列的位置。 基于后缀树的索引Suffxarrays(Salmon,STAR):能快速解决很多关于字符串的问题。缺点:索引所需空间较大,对内存要求较高。 BWT基于块排序压缩的索引Burrows-WheelerTransform(BWA,Bowtie2): 功能 软件 RNA-seq比对到参考基因组上 STRA、HISAT2、Bowtie2、TopHat、TopHat2 转录组组装 StringTie、Cufflinks 质控分析和差异表达基因分析 HTSeq、edgeR、DESeq2、Salmon、FastQC(读序质控) 长片段RNA-seq分析 PacBioIso-seq,Spaln,StringTie 染色体分析ATAC-seq/CHIP-seq Bowtie2,MACS2,ChromVAR、Ballgown 基因组浏览软件 JBrowse RNA-seq数据是cDNA测序(mRNAs剪接之后)。 (1)很多读序来自于相邻的两个或多个外显子。怎样才能把这些读序成功地映射(Mapping)到基因组上? (2)由于读长很短,如何把他们组装成为全长转录本,进而组装成一个转录组? (3)如何来对转录本或基因的表达水平进行定量? 读序映射、转录本组装和表达定量是RNA-seq数据分析紧密相关的三个步骤。 读序映射mapping:映射到转录组或基因组上,其实是一种序列比对的问题。 转录组重构:把大量读序组装成各种各样的转录本进而反映一种细胞类型转录组的问题。 转录表达分析:首要问题就是如何消除系统变异,不能用简单的读序数量来表示一个转录本的丰度,通常采用RPKM(readsperkilobaseoftranscriptpermillionmappedreads),对于双端测序来说,一对读序代表一个片段,因此定量单位变成了FPKM(fragmentsperkilobaseoftranscriptpermillionmappedreads)。 把读序分配给共享外显子的转录本:根据一个基因的所有外显子上读序或者一个基因所有转录本共享外显子上的读序的数量来进行定量。 DNA和组蛋白组装成染色质,DNA和各种酶(DNA、RNA合成酶,DNA切割酶等)以及各种调控蛋白质相互作用。这些蛋白质会定位在基因组的什么位置就是序列富集分析。 有哪些分析技术: TF的识别:DBDbyPfam,SMART,InterPro/HMM TFBS的识别: Gelshift(Electrophoreticmobilityshiftassay,EMSA) DNAfootprint SELEX(SystematicEvolutionofLigandsbyExponentialEnrichment) Proteinbindingarray ChIP-based DamID-seq ChIP指染色质免疫共沉淀技术,seq指的是二代测序方法,chip-seq可以识别蛋白质与DNA互相作用情况。 原理步骤: 原理:在抗体引导下,ChiTag酶仅在目的组蛋白修饰标志、转录因子或染色质调控蛋白结合染色质的局部进行目的DNA的片段化,同时添加测序接头,并释放到细胞外。 CUT&Tag主要特点: ATAC-seq原理: MNase-seq、DNase-seq、TACA-seq、三个的区别如下: 一级结构:核小体结构。由核心颗粒和连接部构成。核心颗粒是一个由H2A、H2B、H3、H4各一对组成的八聚体蛋白,形似算盘珠,DNA分子以140个碱基对长度在其表面缠绕。连接部由60个碱基对的DNA链和一个H1分子组成。核小体的横径约10nm,高6nm,呈扁圆形的球状体。一串一串的核小体形成了直径10nm,电镜下清晰可见的细丝。 二级结构:螺线管。核小体串珠围绕一个空心轴,螺旋化形成外径30nm,内径10nm,螺旋间距11nm的中空线状结构,每一螺旋由6个核小体组成,组蛋白H1位于螺线管内侧,称之为螺线管,即为染色质的二级结构。 三级结构:超螺线管结构,即染色单体。 四级结构:明显的看到微带和两条姐妹染色单体结构。 是染色质构象捕获技术,用于研究基因的不同状态,用于点对点的基因互作,即特定的两个基因间的互作。 主要方式是通过3C甲醛交联固定蛋白与DNA,然后限制性内切酶进行酶切破碎,重新连接交联DNA片段 的粘性末端。互作DNA的两端具有相同的粘性末端,可以互相连接形成loop,采用人工重连确认片段来 自基因组的某个位置。 在研究一个基因与多个基因互作,或多个基因与多个基因互作时就引入了4C,5C,Hi-C技术。 以整个细胞核为研究对象,研究全基因组范围内整个染色质DNA在空间位置上的关系,通过对染色质内全 部DNA相互作用模式进行捕获,获得高分辨率的染色质三维结构信息。 染色质构象捕获技术——3C、4C、5C、Hi-C RNA与染色质互作捕获技术——GRID-seq,通过深度测序实现全球RNA与DNA的相互作用 染色质互作数据分析工具——HiC-pro、GRID-tools 1.转录组数据分析:数据整合的基本思路,微阵列的两大类,BeadChip的基本思想和解码过程(看那个图),归一化的基本概念,QuantileNormalization算法实现 2.基因差异表达分析:差异表达分析的统计学原理,几种基本算法,NewTuxedo套件进行转录组差异表达分析的基本步骤。(mapping,定量,差异,作图,那个组件做什么事情) 3.聚类分析和主成份分析:层次聚类的基本思想和算法实现,分割聚类的基本思想和算法,主成分分析的基本思想(算法思想) 4.单细胞转录组测序分析:实验的基本步骤,现有缺陷,分析的基本步骤,UMI的原理(重要的地方)。 5.单细胞表观组学及多组学整合分析:单细胞表观组主要技术,空间转录组主要编码方式 6.基因调控网络的基本概念:基本概念(哪几部分组成,数学模型,几种形式),基因调控网络的主要构成,模型的几种主要形式。 特征选择:利用先验的知识或基于一定的假设来直接选取一些特征。 特征提取:利用主成分分析(PCA)等方法把数据从高维空间投射到低维空间。相关的方法还包括K-means聚类、等级聚类等 (1)基于知识的方法。利用外部信息等各种知识库,进行富集分析,检测不同组学分析得到的生物实体集合是否富集特定的功能注释。把多组学结果投影到一个事先利用各种知识构建的代谢网络模型中。 (2)数据驱动的方法。利用统计模型和机器学习方法来获得生物实体在多组学数据层内和层间的关系。可以进行分步整合,先分析单独的组学数据或它们的一些简单组合,然后再把结果整合到一起。也可以进行同时整合,将所有的多组学数据集放在一个统一的模型中进行分析。 (3)复合网络的方法。利用基于知识和数据驱动的两种方法获得的不同组学分析结果整合到一个异质性的网络模型中。 4、整合后的可视化与解释。整合分析得到一个非常复杂的网络,需要对其进行可视化以及具有生物学意义(功能)的解释。 DNA微阵列:通过生化反应来检测生物分子或细胞的存在。 DNA微阵列工作原理: 分类: 基本思想:BeadChip的制造是以固体珠子为载体,上面合成特定序列的探针,然后把各种珠子混在一起铺洒到BeadChip上。BeadChip载体表面有大量的直径与珠子相当的小坑(well),每个小坑只能容纳一个珠子。在铺洒过程中,每个珠子落入一个小坑的过程是随机的。 解码技术: 没有那么多种类的荧光基团,但可以利用有限颜色的多步骤排列来标记每一个解码序列,这样就能通过多步骤的杂交反应来实现探针的解码。比如如果有8种探针,两种颜色,那么三个杂交反应就能实现解码。 归一化的概念:由于随机变异和系统因素会产生观测值变异,消除由于系统变异产生的我们不感兴趣的因素OV的过程就是归一化。 可以利用全局性特征,如所有基因表达值的平均值和中位数,某一种百分位数来进行归一化。 利用不同芯片上所有基因的平均表达值来对任何基因X的表达值进行归一化。在同一张芯片上所有基因的都进行了等比的缩放,这种方法被叫做Scaling法,其数学运算的实质是截距为0的线性回归问题。 对整体数据的不同局部进行线性回归建模,得到总体的非线性的平滑回归曲线。找到一组其表达值的顺序不变的基因,利用loess等方法建立回归曲线,就能对所有数据进行归一化。 QuantileNormalization(QM)是一种解决了scaling方法固有缺陷的运算快速的非线性方法。 它所基于的假设是每个芯片(样品)所有基因表达值的分布是相同的。 它的基本流程如下: 在偏差消除方面(包括运算速度)QM的表现要比其他两种方法更好。 利用scaling和loess进行多芯片的归一化时,人们往往选择其中一个芯片作为基准芯片。这种做法存在改变原始总体数据分布的不足之处,这是因为选择所谓的基准芯片实质上是没有充分利用所有芯片数据。 QM充分利用了所以芯片数据,所以能较好地解决这一问题。 另一种解决方法是,利用多个芯片的mean或median值产生所谓的synthetic基准芯片。 RNA-seq中归一化方法就是把读序数转化为RPKM或FPKM。 RPKM(readsperkilobaseoftranscriptpermillionmappedreads) FPKM(fragmentsperkilobaseoftranscriptpermillionmappedreads)。 对差异表达基因检出的ROC曲线来评估来看,upper-quartile上四分位数的表现最优。 当差异基因的表达量较高时,FPKM比QM好; 当差异基因的表达量较低时QM比FPKM好。(因为一个样品中总体读序计数主要是由高表达基因贡献的。) 1型错误:原假设是正确的,却拒绝了原假设。 2型错误:原假设是错误的,却没有拒绝原假设。 各种正确率和错误率的计算方法:FDR(falsediscoveryrate))=1-specificity ROC曲线:横坐标是假阳性率,纵坐标是真阳性率。TPR=TP/TP+FN;FPR=FP/FP+TN t检验:用来比较两组物体的某一属性X是否相同。其实质就是把两组物体的测量值综合成为一个衡量其差异的t统计参数。 t检验的前提条件是变量的概率分布是正态分布。 由于t检验的显著性水平意味着I型错误率。当我们同时研究10,000个基因时,p=0.01就意味着我们会(最多)有100假阳性差异表达基因的存在。 既能给出差异表值,也能给出差异表达基因的数量。给出FDP值评判显著性。该方法基于这样一个实验设计:(U1A,U1B,U2A,U2B,I1A,I1B,I2A,andI2B)实验有8套芯片数据:U代表对照组,I代表处理组(离子辐射处理),1和2代表两种不同的细胞系,A,B代表样品的两个技术重复(同一个样品有两个芯片检测数据)。 一般使用FoldChange(FC)值来确定DEG。RP方法既有统计学依据又符合生物学家对FC之喜好的方法。这个方法既有FC法的简单直观,又克服了其缺乏统计学判断的缺陷。 RP在以下几方面比SAM的表现要更好:(1)同一基因的不同探针对的重复性更好:(2)对生物重复的依赖性更低。使得RP有更好表现的原因有以下几点:对数据的预设条件比较宽松,比如它并不要求数据的正态分布特性;利用基因的FC来作为一种特征变量具有更直观的生物学意义。也就是说,变化倍数越大的基因越可能是关键基因,其统计学的显著性也越强。RP的统计参数更加稳定,变异更小,对生物重复要求低。 RP是一个合理、直观、经济、简单的DEG分析方法,它可以降低研究的成本,可以被用于芯片以外的其他技术平台,如质谱数据分析等。 (1)核酸杂交固有的高背景值;(2)不同探针的检测能力差别很大(探针效应);(3)无法对不同基因或转录本的表达进行比较;(4)不能发现新的转录本。 RNA-seq数据分析的实质是计数数据(countdata)的分析。其面临的挑战包括:(1)技术数据的非正态分布;(2)计数数据变异对平均值的依赖性;(3)低生物重复数(一个条件一般只有2-3个生物重复) 开始人们认为读序计数(readcounts)接近泊松分布,但是后来发现泊松分布所预测的变异比实际小,因此导致了过高的TypeIerror。人们发现,RNA-seq的readcounts更符合负二项式分布。 NewTuxedo进行转录组差异表达分析的基本步骤。 NewTuxedo由HISAT和StringTie构成。 聚类分析:是一种将物体分组的方法,其结果是同一组内物体的相似度要高于来自不同组的物体的相似度。是一种无监督的机器学习方法。 聚类分析的第一步是要确定衡量两个物体相似度或差异度的一种度量方法。也就是要从原始的数据矩阵获得一个差异矩阵。在把数据矩阵转变成差异矩阵之前,需要把数据进行归一化。最普遍的差异度量是欧氏距离。 层次聚类的基本思想:是按照一个树状结构来把物体分类。树的叶节点是一个个物体(可以看做只包含一个物体的类群)。取决于等级构建是从下到上还是从上到下,等级聚类分成聚合型和分割型两类。 操作过程: 1.启动:把每一个物体看作一个类; 2.循环:找到两个最相似的类群;把它们合并成一个类; 3.停止:当类的数目达到预设值时停止。 两个类群的相似度:是它们所包含的物体间的相似度的某种取值,可以有三种方法: •完全链接法:把物体间的最大距离当做两类间的距离; •平均链接法:把物体间的平均距离当做两类间的距离; •单一链接法:把物体间的最小距离当做两类间的距离。 完全链接法形成的类比较紧凑,单一链接法形成的类比较细长。平均链接法形成的类群的形状居于前二者之间。 分割聚类的基本思想:是把n个物体分成k个类群,使得物体在类群内的差异小于类群间的差异。等级聚类的做法是把1≤k≤n的所有k都做了一遍,而分割聚类只做一个特定的k值。分割聚类需要预先设置一个目标目标函数,然后逐步对其进行优化。因此,分割聚类是一种优化算法。目标函数一般就是组内物体的差异度。 K-means聚类算法就是分割聚类,其步骤如下: 1.启动:随机选取属性空间的K个点作为k个类群的中心点ci; 2.循环:1).根据每个点p与ci的距离把他们分到相应的簇Ci,即把p分配到与其距离最短的ci代表的Ci(c代表中点,C代表类群);2).计算每一个Ci的平均值,把平均值作为新的ci; 3.停止:当每一个点p的分类不再变化时停止。 主成分分析PCA:是一种用于降低数据维度的统计学方法。 通过降维我们可以去除数据的冗余,实现数据的压缩。 降维不是简单的删除一些维度,而是将原来的高维空间转化为新的低维空间,将物体(数据点)从一个高维空间映射到一个新的低维空间。 PCA的两个基本思想: Principle1:两个变量的高度相关性是它们数据高度冗余性的特征; Principle2:一个变量的变异越大说明它越能反映底层机制的动态过程。 PCA要做的就是把原始数据改写成另一个属性空间里的新数据,新数据保留了旧数据的所有信息而去除了冗余和噪音。 PCA是一个分析高维度数据的一个有力工具。需要满足四个主要的条件: 1.线性化高维数据和低维数据之间的变换是线性的。或者说,不同检测数据是源头信号的线性组合。 2.变异越大的维度越是我们感兴趣的属性。 3.均值和方差是描述数据的充分条件。如果数据不符合正态分布,则不能用PCA。 4.信号源对数据的产生是独立的。 设有m条n维数据。 1)将原始数据按列组成n行m列矩阵X 2)将X的每一行(代表一个属性字段)进行零均值化,即减去这一行的均值 3)求出协方差矩阵 4)求出协方差矩阵的特征值及对应的特征向量r 5)将特征向量按对应特征值大小从上到下按行排列成矩阵,取前k行组成矩阵P 6)即为降维到k维后的数据 1.单细胞的制备。可以通过稀释、口吸管挑取、流式细胞仪分选、激光切割、微流控捕获。基于液滴的技术和Mircowell的技术由于具有快速获得大量细胞的优势,适合用于研究复杂组织中的细胞异质性。 大多数采用油包水技术,将一个磁珠和一个细胞包括在同一滴油中,然后再油滴里进行细胞裂解反应,裂解后序列被磁珠吸附。Barcode区分不同的细胞,umi区分不同的序列。 2.细胞的裂解。利用温和的裂解液以免对下一步造成不良影响。 3.反转录。尽量反转录出更长的cDNA就能够提高转录本的覆盖度,也就能提高scRNA-seq的检出率。 4.DNA扩增。DNA的非线性PCR扩增是造成scRNA-seq准确性差的主要原因 5.转录本的覆盖。有的读长覆盖整个转录本。 细胞内RNA的降解导致有些细胞的读序数非常低; 能测到的有表达的基因数很低; 因为线粒体有自己的膜,其RNA的降解会最后发生,因此线粒体表达基因数高; 由于要分批做,由于操作者、试剂和测序仪引起批次效应; 表达值的计算,归一化后计算表达值。 数据可视化。 降维分析。 伪时间轨迹分析。 由于反转录后形成的cDNA需要被PCR扩增,而PCR扩增并非线性的,所以导致最后的表达值(比如RPM)不能正确反映扩增前cDNA之间的比例。 为了解决这一问题,可以把扩增前的每一个cDNA分子都做上一个umi标签。 在扩增前预先把cDNA分子和UMI序列连接,就相当于我们把某个cDNA的多个不同的分子贴上了不同标签(事实上UMI是在合成第二条cDNA链时被加上去的)。这样,在PCR放大和测序完成后,不管细胞的读序数有多少,我们都把它测到的标签数作为表达值。对于其cDNA也是如此。 这样通过测序得到的表达值不仅保持了cDNA原来分子数的比例,还可以作为一种绝对定量的方法。 UMI不能用于全长转录本测序。 对细胞进行质控质控指标包括: sum:每个细胞的读序总数. detected:每个细胞检测到的基因(特征数)数目(读序数>0)。 subsets_X_percent:每个细胞对照特征集X(比如线粒体基因)读序数百分比。 对基因进行质控:质控指标包括: mean:每个基因/特征在所有细胞上的平均读序数; detected:每个基因读序数不为0的细胞的百分比; subsets_Y_percent:每个基因的对照细胞集Y(比如没有细胞的对照样品测序)读序数在所有细胞读序数中的百分比。 由于操作者、仪器、试剂不同会产生所谓的批次效应。 在高纬度空间寻找mutualnearestneighbors(MNNs)的方法来更好地解决批次效应。 MNNs主要思想是在两个不同批次或同一批次的两个重复样品中鉴定表达谱相互相似的细胞,并假设这些细胞在基因表达的高纬度空间的不同位置是由 批次效应引起的。在消除批次效应后不同批次的数据可以整合在一起形成一个数据集。 步骤: 玻片位置编码:玻片是一个个喷墨的小个子,每个玻片上喷上oligo,将样品放在玻片上,就知道当前组织的细胞的位置。然后测序,测序后每个基因带有玻片的位置编码,与原来玻片的位置对照,找出重叠高亮部分,表明这个基因在该位置高表达。 荧光序列编码:对每个基因有不同的荧光颜色编码,在不同时期在玻片上进行不同的反应,得到很多个不同的荧光图像,将多个荧光图像重叠就可以得到不同基因表达的位置。 微流控编码:将组织放到玻片上,将一个有多个管道barcode盖到玻片上,不同的通道根据x轴和y轴进行编码,得到一个类似马赛克图的结果。 由调控基因、DNA调控元件及其相互作用组成的系统,决定基因表达的时间和空间。 ODE常微分方程。精确描述;所需参数极多,几乎不现实 BayesianNetwork贝叶斯网络。所需数据较多,只适用于模建小型网络 BooleanNetwork布尔网络。易于理解和计算;定性描述 Implicitmodeling隐式建模。易于理解;定性描述 ANANSE:Bulkmulti-omics SCENIC:scRNA-seq+TFBSaroundTSS GRNsmile:scRNA-seq+scATAC-seq+Micro-C+TFBS+Datavis 1.深度学习的计算基础:机器学习的计算原理,感知机的关键特征,多层感知机的节点层 2.卷积神经网络与图像识别:反向传播算法、卷积运算的基本概念 3.生物影像组数据的深度处理:卷积神经网络的常用框架,卷积神经网络AlexNet的关键特征 4.神经网络的应用与局限性:序列神经网络的特点,VNN、RNN、LSTM等模型等基本概念 机器学习:是一类算法的总称,这些算法企图从大量历史数据中挖掘出其中隐含的规律,并用于预测或者分类,更具体的说,机器学习可以看作是寻找一个函数,输入是样本数据,输出是期望的结果,只是这个函数过于复杂,以至于不太方便形式化表达。 K邻近算法KNN,决策树DT,随机森林RF,朴素贝叶斯,人工神经网络,BP,支持向量机SVM,多种回归 模型(logistic回归算法,线性回归,生存分析)梯度提升树GBDT,卷积神经网络CNN,LSTM,GAN。 K-MEANS聚类,稀疏自编码,限制波尔兹曼机,高斯混合模型,主成分分析,系统聚类,FCN,DBASCAN等 一个简单的感知机是将每个输入设置相应的权重,将所有输入加权求和到神经元上,每个神经元都有自己的权重和偏置。神经元的输出作为激活函数的输入,激活函数的结果作为最后的输出。 一个输入层,一个隐藏层,一个输出层。 在单层神经网络的基础上引入了一到多个隐藏层。隐藏层位于输入层和输出层之间。隐藏层具有不同数量的神经元,每个隐藏层神经元具有相同的激活函数。不同层之间是全连接的。 在神经元中,输入的值通过加权求和后,还被作用了一个函数,这个函数就是激活函数。 激活函数将神经元的输入映射到输出端。将非线性特性引入到我们的网络中。 如果不用激活函数,每一层输出都是上层输入的线性函数,无论神经网络有多少层,输出都是输入的线性组合,这种情况就是最原始的感知机。 激活函数都是非线性的。 BP算法的学习过程由正向传播过程和反向传播过程组成。在正向传播过程中,输入信息通过输入层经隐含层,逐层处理并传向输出层。如果在输出层得不到期望的输出值,则取输出与期望的误差的平方和作为目标函数,转入反向传播,逐层求出目标函数对各神经元权值的偏导数,构成目标函数对权值向量的梯量,作为修改权值的依据,网络的学习在权值修改过程中完成。误差达到所期望值时,网络学习结束。 反向传播算法主要由两个环节(激励传播、权重更新)反复循环迭代,直到网络的对输入的响应达到预定的目标范围为止。 激励传播:每次迭代中的传播环节包含两步:(前向传播阶段)将训练输入送入网络以获得激励响应; (反向传播阶段)将激励响应同训练输入对应的目标输出求差,从而获得隐层和输出层的响应误差。 权重更新:对于每个突触上的权重。将输入激励和响应误差相乘,从而获得权重的梯度;将这个梯度乘上一个比例并取反后加到权重上。 卷积运算:是指从图像的左上角开始,开一个与模板同样大小的活动窗口,窗口图像与模板像元对应起来相乘再相加,并用计算结果代替窗口中心的像元亮度值。然后,活动窗口向右移动一列,并作同样的运算。以此类推,从左到右、从上到下,即可得到一幅新图像。 CovNet、LeNet、AlexNet、VGG、GoogleNet、ResNet、DenseNet、 AlexNet的提出是基于分组卷积这种思想。 1、在每个卷机后面添加了Relu激活函数,解决了Sigmoid的梯度消失问题,使收敛更快。 2、使用随机丢弃技术(dropout)选择性地忽略训练中的单个神经元,避免模型的过拟合。 3、添加了归一化LRN(局部响应归一化)层,使准确率更高。 4、重叠最大池化,即池化范围z与步长s存在关系z>s避免平均池化的平均效应。 丢弃率:也是经常说的一个概念,能够比较有效地防止神经网络的过拟合。相对于一般如线性模型使用正则的方法来防止模型过拟合,而在神经网络中Dropout通过修改神经网络本身结构来实现。对于某一层神经元,通过定义的概率来随机删除一些神经元,同时保持输入层与输出层神经元的个数不变,然后按照神经网络的学习方法进行参数更新,下一次迭代中,重新随机删除一些神经元,直至训练结束。 深度学习框架: caffe、torch、theano、tensorflow、CNTK、keras 经典的卷积神经网络都是输入和输出一一对应,也就是一个输入得到一个输出。不同的输入之间是没有联系的。但在某些场景下,一个输入不够用,还需要不同的输入之间的序列顺序关系,即相互依赖的数据流, RNN跟传统神经网络最大的区别在于每次都会将前一次的输出结果,带到下一次的隐藏层中,一起训练。 一个简单的RNN模型如下所示,它由输入层、一个隐藏层和一个输出层组成,在全连接神经网络基础上新增一个w,x是输入矩阵/数据,s是隐藏层数据,o是输出层,u是输出层到隐藏层的权重矩阵,v是隐藏层到输出层的权重矩阵,循环神经网络的隐藏层的值s不仅仅取决于当前这次的输入x,还取决于上一次隐藏层的值s。权重矩阵W就是隐藏层上一次的值作为这一次的输入的权重。 优点:它能有效的处理序列数据,在序列中前面的输入也会影响到后面的输出,相当于有了“记忆功能”。 缺点:RNN存在梯度消失的问题,RNN中越晚的输入影响越大,越早的输入影响越小,且无法改变这个逻辑。存在严重的短期记忆问题,长期的数据影响很小(哪怕他是重要的信息)。 了解决循环神经网络无法捕获数据长距离依赖的缺陷,产生了LSTM,其中包含遗忘门、输入门、输出门三个门,而且在不同的门使用不同的激活函数以达到不同的目的。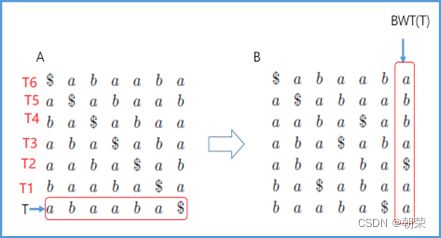
(3)数据库序列搜索的基本思路
(4)Motif基本概念
(5)模件搜索的方法
(6)描述Motif的几种常用方法

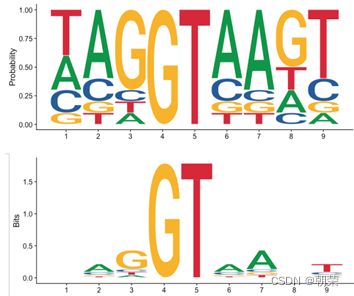
(7)序列库的主要索引方法

(8)NGS序列分析软件
(9)RNA-seq数据分析步骤

(10)序列富集分析技术
(11)DNA调控元件的主要类型
(12)鉴定调控元件的常用实验方法
DNA序列分析:
生物化学注释
eQTLmapping
(13)转录因子和转录因子结合位点的识别
(14)调控元件相关实验方法

1、Chip-seq:
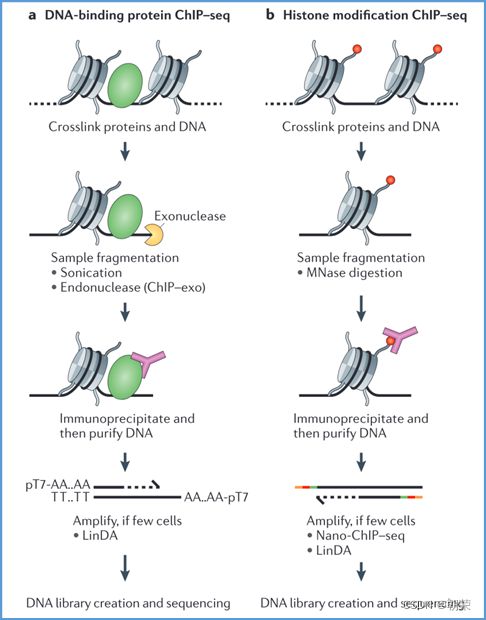
2、CUT&Tag:
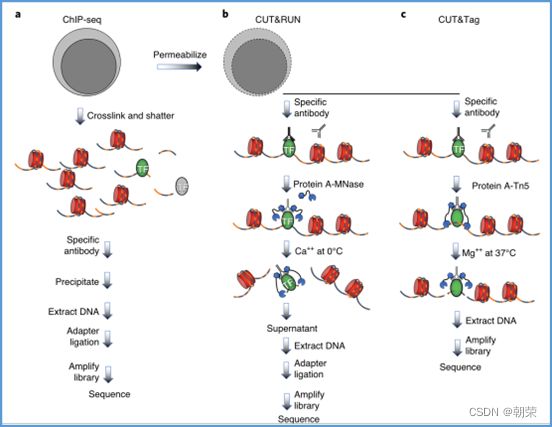
3、ATAC-seq染色质可及性
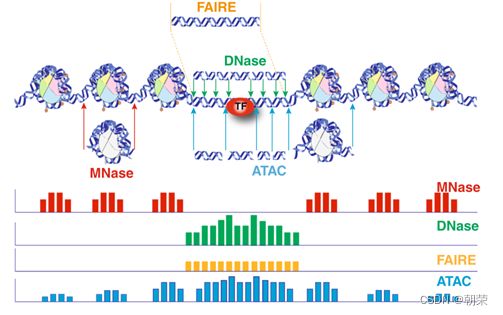
(14)序列关联分析:
染色质构象的基本概念
染色质成分:
染色质结构:
染色质分类:
主要相关实验方法
3C-Seq:
Hi-C:
第三章多组学数据的整合分析
(1)转录组数据分析:
数据整合的基本思路:
(2)微阵列
(3)BeadChip
(4)归一化
1.Scaling:
2.Non-linearmethod非线性方法:
3.QuantileNormalization算法实现
三种归一化方法的评估:
RNA-seq中的归一化:
(5)基因差异表达分析
差异表达分析的统计学原理

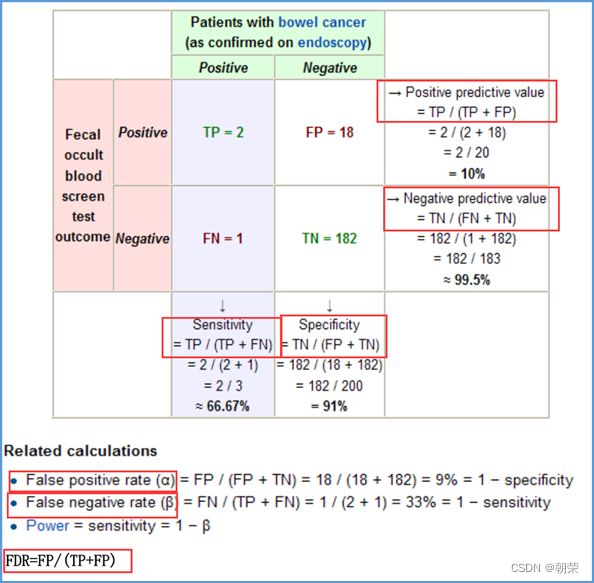
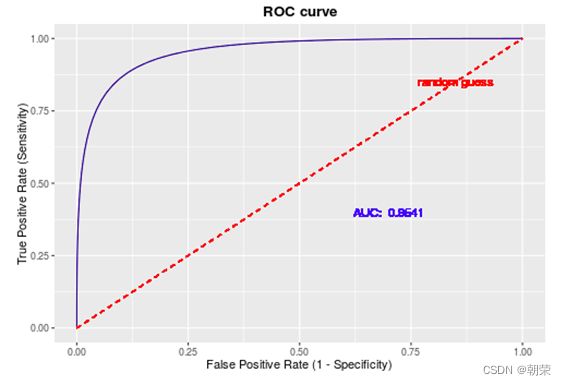
t检验
差异表达分析方法
1.SAM:微阵列的重要分析。
2.RP:RankProducts。
芯片技术固有的缺陷包括:
RNA-seq面临的挑战
(6)NewTuxedo套件差异表达分析
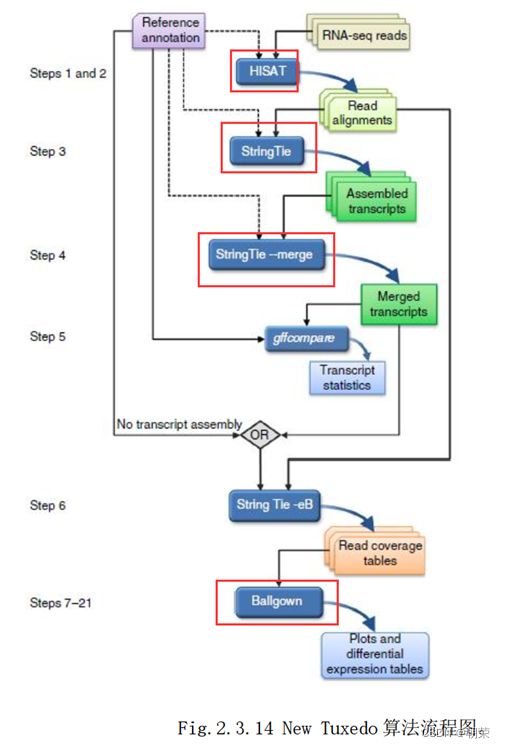
(7)聚类分析和主成份分析
层次聚类的基本思想和算法实现
分割聚类的基本思想和算法
PCA的基本思想
PCA步骤:
(8)单细胞转录组测序分析
实验的基本步骤
scRNA-seq的缺陷:
scRNA-seq分析的基本步骤:
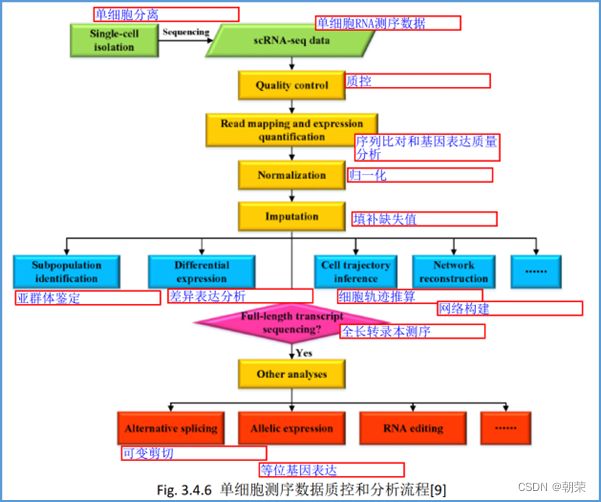
单细胞测序数据分析:
UMI的原理(重要的地方)
质控
单细胞测序数据批次效应处理
(9)单细胞表观组学及多组学整合分析

单细胞表观组主要技术
空间转录组主要编码方式


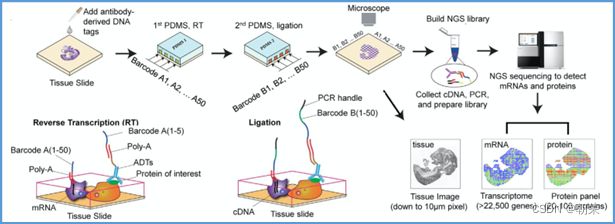
(10)基因调控网络的基本概念

基因调控网络组成:
模型的几种主要形式
GRN基因调控网络解析
第四章多组学数据的深度分析
(1)机器学习的计算原理

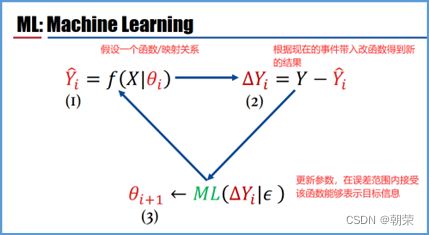

监督学习(当训练样本带有标签时):
非监督学习(训练样本全部无标签时):
(2)感知机的关键特征
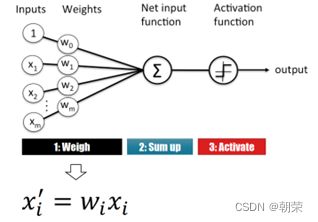
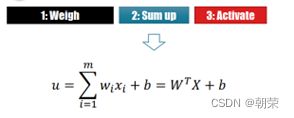

单层感知机:
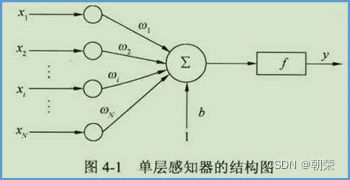
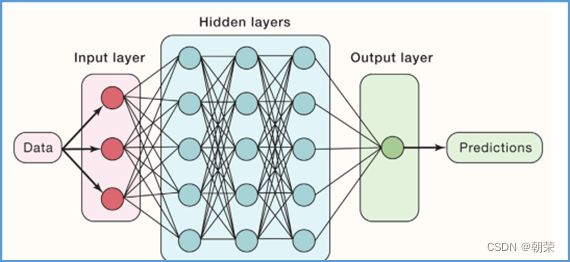
多层感知机:
激活函数

(3)卷积神经网络与图像识别:
反向传播算法
算法原理:
卷积运算的基本概念

(4)生物影像组数据的深度处理:
卷积神经网络的常用框架
卷积神经网络AlexNet的关键特征
AlexNet特点:
Dropout
(5)神经网络的应用与局限性:
序列神经网络的特点
RNN

LSTM长短时记忆神经网络