测序组学助力新的酶发现
2020年,深度学习算法AlphaFold2在从原始序列预测蛋白质三维结构方面取得了里程碑式的成果。
宏基因组学产生的大量测序数据,让人们得已窥见未经培养的微生物的生物合成潜力。与初级代谢途径相比,参与次级代谢的酶往往催化不同底物的特殊反应,这些途径为发现新的酶学提供了丰富的资源。
到目前为止,从环境DNA(eDNA)研究中发现新的酶或功能大多数是通过PCR筛选或基于活性位点的筛选方法获得的。作为另一种选择,鸟枪法宏基因组学也具有从eDNA中直接发现新酶的能力,还可以避免由于PCR或活性导向的功能宏基因组学工作流程引入的共同偏差。
最近发表的一篇长综述,为宏基因组学在酶学领域构建了一张宏伟蓝图。文章中比较了发现酶的方法,包括系统发育学、序列相似性网络、机器学习技术等。也讨论了各种实验策略来测试计算预测,包括异源表达和筛选。
除了这些广泛使用的方法,还补充了一些新兴技术如宏组学、单细胞基因组学、无细胞表达系统等方法及建议。这里,我们沿着作者给出的路线,为大家做个导读。
首先作者在文章中明确指出两点,文章中主要关注天然产生的酶,而不包括通过工程或定向进化策略获取的非自然酶。其次是生物合成基因簇(bgc)中编码的细菌酶,因为这些酶是天然产物中研究最广泛的。
另一点宏基因组DNA序列与从微生物分离物中获得的基因组DNA没有本质上的区别。两者都是来自生物系统的核苷酸序列。从结构上讲,宏基因组样本中的BGCs与分离物参考基因组中的BGCs基本上没有区别,除了有时由于组装过程中引入的相邻边界和错误而更碎片化。一些宏基因组BGCs甚至在可培养生物的基因组中具有同源簇。
发现新酶的定义
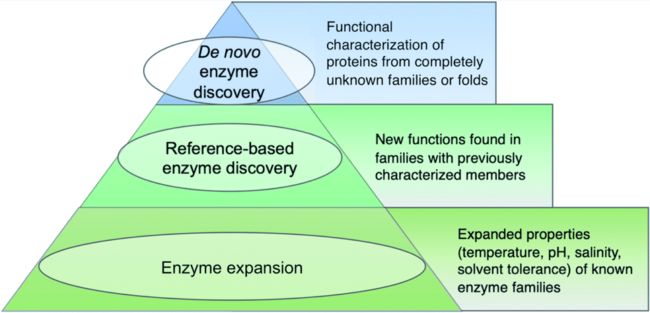
如下图,使用三层金字塔说明,越往下说明这个类别在宏基因组酶研究中数量更多。
Robinson S Let al., Nat. Prod. Rep., 2021
第一层
即金字塔尖端,指的是识别出全新类型的生物催化剂,也就是说这类酶必须属于没有任何功能特征成员的蛋白质折叠或家族。到目前为止,大多数新发现的酶的例子都来自可培养的细菌和真菌,而不是eDNA和未培养的微生物。也因此,在宏基因组中识别出的蛋白质家族中还存在极大的探索空间。
第二层
指的是基于参考发现的酶,是在已发现的蛋白质家族中对新的反应类型的表征。
第三层
代表了宏基因组酶研究中占比最大的一部分,指发现了具有不同底物种类的酶,或具有不同反应条件的酶,包括温度、pH、盐度或溶剂偏好。
宏基因组学研究的实验设计
在发现酶的方法中,将鸟枪法宏基因组学测序与功能宏基因组学(活性导向分离和基于PCR方法)之间进行比较。

Robinson S Let al., Nat. Prod. Rep., 2021
活性导向分离方法筛选功能宏基因组文库是宏基因组领域最早发展起来的方法之一,方法核心是鉴定出所需表型的克隆,例如从fosmid、cosmid或人工染色体文库克隆。由于该工作流程不依靠序列同源性,因此对从头发现新酶特别有效。
基于PCR方法的筛选核心是简并引物以扩增编码感兴趣的蛋白质结构域的eDNA基因。基于扩增的常见的生物合成标记物的分析已经被广泛地应用于检测新的BGCs和天然产物。例如,一类全新的钙依赖性抗生素,苹果酸,是通过基于PCR的土壤亚基因组腺苷酸结构域筛选检测到的。
鸟枪法宏基因组学是指直接的、非靶向的eDNA测序。由于不需要PCR扩增和大肠杆菌等文库宿主,所以在鸟枪法测序过程中引入的偏差较少。产生测序数据的速度比构建宏基因组fosmid或cosmid文库快得多。其最大的挑战是从复杂环境样本中足够数量和质量的eDNA和足够的测序深度来检测和纠正个别读数中的错误。关于检测稀有生物的BGCs,可以使用Samplix技术。
这一小节重点介绍了发现酶的三种方法,虽然各有参差,但是殊途同归,依靠这些技术新的酶不断被发现。不容忽视的是应用于鸟枪法宏基因组测序数据的生物信息算法和技术的进步为酶的发现提供了新的途径。但是参与天然产物生物合成的酶是如何帮助从宏基因组数据集中获得要点,以提高我们对未培养微生物的次级代谢功能的认识呢?作者提出一个问题,“是否存在发现酶的温床?”
酶功能预测的计算方法
鸟枪法宏基因组测序完成后,就需要执行下游生物信息分析,使数据可公开存取使用,例如JGI IMG/M、iMicrobe或MGnify这些站点,整合了大量的基因组数据,可以分析可以存储。
这里作者特别介绍了MGnify,MGnify的制作作者强调它是为了“搜索微生物暗物质”而开发的。MGnify的一个好处是能够使用HMMs查询宏基因组,而不是使用基本的基于序列比对的搜索方法,如BLAST或DIAMOND。
虽然这两种方法都是有效且快速的方法,但HMMs对于鉴定更遥远的同源基因特别有用。
(MGnify:https://www.ebi.ac.uk/metagenomics/)
预测蛋白质家族中新的酶功能的计算方法之间的比较

Robinson S Let al., Nat. Prod. Rep., 2021
基于以上的计算方法,整理了作者在文章中列举的一些常用工具:

Robinson S Let al., Nat. Prod. Rep., 2021
实验策略:表征新的酶
无论是用鸟枪法还是功能筛选的宏基因组学发现酶,最后都需要对酶进行表征。
1 质量控制
当选择蛋白质在实验室中进行鉴定时,重要的第一步是质量控制,以去除可能存在测序错误或不能编码全功能蛋白质的嵌合体和截断序列(truncated sequences)。可以根据相似性对蛋白质进行聚类,并自动选择有代表性的序列,比如CD-HIT和UCLUST工具。
根据数据集的大小,可能需要进一步的过滤步骤。
最明显的策略之一是选择在可培养生物体中也存在的宏基因组序列,因为这可以在原生宿主中进行功能表征。其次是从嗜热生物体中选择蛋白质,这些蛋白质往往编码热稳定性更高的酶。还有选择更稳定和表达更好的蛋白质,包括过滤不具有高GC含量、跨膜区或无序区的蛋白质。
作者建议使用多种标准来对需要实验鉴定的蛋白质序列进行排序,通过这种方式,预测工具中的个体偏差可能会被基于集成的方法部分抵消,以确定最有希望的能够表征发现的酶的蛋白质。
2 蛋白异源表达
一旦识别了感兴趣的酶或BGCs,必须设计异源表达的构建。不幸的是,大多数用于功能宏基因组学方法的宏基因组文库准备的载体通常不适用于异源表达。由于Fosmid/Cosmid载体的最大插入大小为45 kb,许多完整的BGCs也没能完全被捕获到宏基因组文库中。
除了经典的限制性内切酶克隆和Gibson组装方法外,人们还开发了新的方法来提高将大型BGCs克隆到异源宿主的效率和方便性。
一种流行的方法是转化偶联重组技术(TAR),它利用酵母中的同源重组系统将土壤和海绵宏基因组中重叠的eDNA cosmid/fosmid克隆拼接在一起。
3 酶活性的筛选
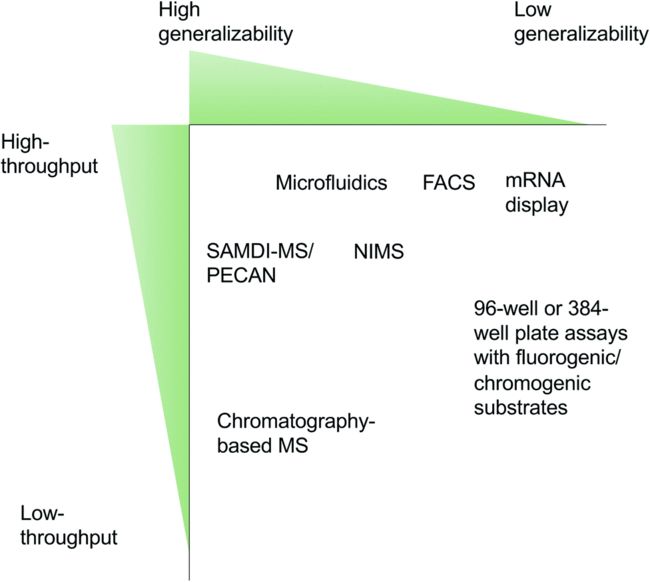
当感兴趣的酶被表达出来后,就要对它们进行体内或体外的活性分析。酶筛选方法通常在通量(throughput)和通用性(generalizability)之间进行权衡,如下图:FACS(流式细胞荧光分选技术)、NIMS(纳米结构启动质谱技术)、SAMDI-MS[ 结合无细胞蛋白质合成和自组装单层解吸电离(SAMDI)质谱技术]、Microfluidics(微流控技术)、mRNA display(通过体外核糖体翻译,有效地将肽链到自己编码的RNA)。
Robinson S Let al., Nat. Prod. Rep., 2021
展望:发现酶的新领域
对该领域的未来提供一个展望,着重于新兴技术与宏基因组学工作流程相结合,以加速酶的发现。
1 宏组学
将各种宏组学技术(包括宏转录组学、宏蛋白质组学和代谢组学)整合到酶发现工作流程中,可以成为一个强大的框架,将基因型与表型联系起来,以产生假说。例如用RNA-Seq分析了一种未知的钼依赖酶DADH在人体肠道中参与多巴胺分解代谢的过程;一项堆肥微生物群落的宏转录组分析结果发现了糖苷水解酶家族中的一个异常酶,这个酶带有exo-1,4-b-xylanase活性等。不同的多组学数据集的整合为酶的发现提供了新途径。
2 单细胞基因组学
单细胞基因组学依赖于微生物细胞的分选,通常采用微流控技术或流式细胞仪(FACS)的方法,然后用高保真聚合酶裂解和全基因组多重置换扩增(MDA)。单细胞基因组学并不依靠于相似细胞的种群是无性繁殖的假设。
因此,单细胞基因组学研究揭示了从海洋浮游植物到癌细胞的各种系统中显著的种群内基因组变异和进化。这一新兴的研究领域需要进一步应用单细胞和空间转录组方法,以更好地了解微生物群落结构和微环境如何影响生物合成基因的表达。
3 微流控
基于微流控的分选方法已被广泛应用于定向进化和蛋白质工程研究,但很少用于挖掘基因组引导酶的发现。最近的一项研究使用光学镊子和微流控技术,根据单个细胞的拉曼光谱对复杂的微生物群落进行分类,这在下游单细胞测序或培养工作中有许多应用。通过对分选的细胞进行下游单细胞测序,活的单个细胞的化学表型可以直接与它们的基因型联系在一起。只是,微流控技术在从宏基因组中发现新的生物合成酶方面的应用目前还没有广泛使用。
4 无细胞系统
无细胞系统为所需DNA序列的快速转录和翻译创造条件,而不受维持细胞生长的限制。与体内表达系统不同,无细胞平台还允许产生有毒的代谢物,这些代谢物通常会杀死异种宿主。为了进一步提高产量,包括mRNA display、MALDI-MS和液滴微流控等筛选方法已经与无细胞平台相结合。对于一些生物合成途径,DNA模板在短短几个小时内就能产生高产量。
5 与序列无关的方法
文中描述的绝大多数技术都依靠基于序列或基于结构的同源性来推断蛋白质功能。然而,当预测“未知的未知因素”时,这些方法往往达不到预期,即重新发现与一个或多个特征蛋白家族没有序列或结构相似性的酶。与序列或结构无关的方法在天然产物研究中也很少使用,因为大多数识别BGCs的计算方法都依靠与常见生物合成结构域的同源性。
decRiPPter是一种基因组挖掘工具,用于检测新的RiPPs和BGCs。decRiPPter算法的核心过滤步骤是使用泛基因组比较来检测分布在分类群内的操纵子,这些操纵子可能参与了次级代谢功能,而不是初级代谢功能。Krousterman等人用DecRiPter分析了1295个链霉菌基因组,鉴定了一个新的RIPP成熟酶家族,催化一种新的肽类天然产物的脱水和环化反应。
基于过往的研究分析的建议
1 新的发现往往发生在蛋白质家族的近邻
虽然这不是一个普遍规律,但与已知功能的参考蛋白相比,序列同源性低的蛋白质比序列同源性高的酶更容易适应不同的底物,并催化出新的反应类型。
2 跳出比色测定法的框框,进入未知的蛋白质空间
对2014年1月至2017年3月发现的宏基因组酶进行的荟萃分析发现,>84%属于脂肪酶/酯酶或纤维素酶/半纤维素酶类别。同样,>82%是通过基于活性的筛选发现的。显然,目前的宏基因组筛选方法偏向于工业相关的酶类,这些酶类也可以用标准比色法检测出来。
3 不再局限于大肠杆菌,寻找新的宿主
一项对照研究发现,一般环境细菌中只有30-40%的基因可以在大肠杆菌中表达,只有7%的高GC含量的DNA可以在大肠杆菌中表达。在功能宏基因组学方面,假单胞菌、链霉菌、红球菌、芽孢杆菌甚至古生菌已经被用作文库宿主和具有穿梭载体的多宿主表达系统(multi-host expression systems)。同样,非传统的异源表达宿主(如亚硝型分枝杆菌)已经被开发用于从宏基因组BGCs发现新的酶。
相关阅读:
生物系统和疾病的多组学数据整合考虑和研究设计
MetaGEM:直接从宏基因组重建基因组规模的代谢模型
ResistoXplorer——基于Web的耐药基因组数据可视化,统计和探索新分析工具
参考文献:
Robinson S L, Piel J, Sunagawa S. A roadmap for metagenomic enzyme discovery[J]. Natural Product Reports, 2021.
E. J. Culp, N. Waglechner, W. Wang, A. A. Fiebig-Comyn,Y.-P. Hsu, K. Koteva, D. Sychantha, B. K. Coombes,M. S. Van Nieuwenhze, Y. V. Brun and G. D. Wright,Nature, 2020, 578, 582–587
N. S´elem-Mojica, C. Aguilar, K. Guti´errez-Garc´ıa,C. E. Mart´ınez-Guerrero and F. Barona-G´omez, Microb.Genomics, 2019, 5, 445270
M. G. Chevrette, K. Guti´errez-Garc´ıa, N. Selem-Mojica,C. Aguilar-Mart´ınez, A. Yanez-Olvera, H. E. Ramos- ˜Aboites, P. A. Hoskisson and F. Barona-G´omez, Nat. Prod.Rep., 2020, 37, 566–599.