SCS【16】从肿瘤单细胞RNA-Seq数据中推断拷贝数变化 (inferCNV)

点击关注,桓峰基因
今天来说说 从肿瘤单细胞RNA-Seq数据中推断拷贝数变化,学会 这些分析结果,距离发文章就只差样本的选择了,有创新性的样本将成为文章的亮点,并不是分析内容了!
单细胞生信分析教程
桓峰基因公众号推出单细胞生信分析教程并配有视频在线教程,目前整理出来的相关教程目录如下:
Topic 6. 克隆进化之 Canopy
Topic 7. 克隆进化之 Cardelino
Topic 8. 克隆进化之 RobustClone
SCS【1】今天开启单细胞之旅,述说单细胞测序的前世今生
SCS【2】单细胞转录组 之 cellranger
SCS【3】单细胞转录组数据 GEO 下载及读取
SCS【4】单细胞转录组数据可视化分析 (Seurat 4.0)
SCS【5】单细胞转录组数据可视化分析 (scater)
SCS【6】单细胞转录组之细胞类型自动注释 (SingleR)
SCS【7】单细胞转录组之轨迹分析 (Monocle 3) 聚类、分类和计数细胞
SCS【8】单细胞转录组之筛选标记基因 (Monocle 3)
SCS【9】单细胞转录组之构建细胞轨迹 (Monocle 3)
SCS【10】单细胞转录组之差异表达分析 (Monocle 3)
SCS【11】单细胞ATAC-seq 可视化分析 (Cicero)
SCS【12】单细胞转录组之评估不同单细胞亚群的分化潜能 (Cytotrace)
SCS【13】单细胞转录组之识别细胞对“基因集”的响应 (AUCell)
SCS【14】单细胞调节网络推理和聚类 (SCENIC)
SCS【15】细胞交互:受体-配体及其相互作用的细胞通讯数据库 (CellPhoneDB)
前 言
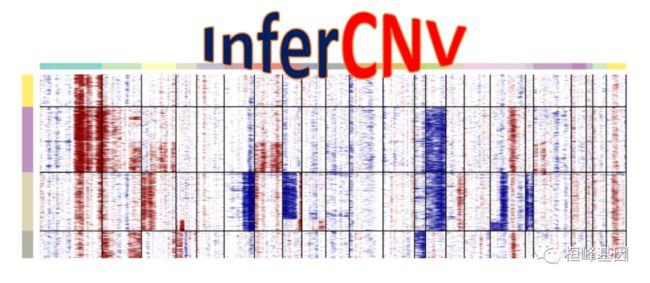
InferCNV用于探索肿瘤单细胞RNA-Seq数据,以确定体细胞大规模染色体拷贝数改变的证据,如整个染色体或染色体的大片段的增加或删除。这是通过与一组参考“正常”细胞相比,探索肿瘤基因组不同位置的基因表达强度来实现的。热图可以显示出每条染色体的相对表达强度,与正常细胞相比,肿瘤基因组的哪些区域过丰或过少,这通常很容易就能看出。InferCNV提供了几个剩余表达式过滤器的访问,以探索最小化噪声和进一步揭示支持CNA的信号。此外,推断cnv包括预测CNA区域和根据异质性模式定义细胞集群的方法。InferCNV是trinitycat工具包的一个组成部分,专注于利用RNA-Seq更好地理解癌症转录组。
分析流程
InferCNV可以用于肿瘤单细胞RNA-Seq数据中鉴定大规模染色体拷贝数变异,例如整个染色体或大片段染色体的扩增或缺失。基本思路是在整个基因组范围内,将每个肿瘤细胞基因表达与平均表达或“正常”参考细胞基因表达对比,确定其表达强度。

从算法的角度上看其实需要四部:
smoothing by chromosome
centering by cell
subtracting normal from tumor cells
de-noising and CNV prediction
我比较关注最后两步,去噪和CNV预测是分开的,一些已发表的文章这部分的分析都是基于去噪之后的结果(比如那张热图),CNV预测结果用的比较少,个人觉得使用analysis_mode = "subclusters"模式后的热图更好看,而且只有6个值这是官网的一张对比图:

软件安装
在安装inferCNV软件包之前我们需要安装JAGS,window版的直接下载.exe即可,下载链接:
https://jaist.dl.sourceforge.net/project/mcmc-jags/JAGS/4.x/Windows/JAGS-4.3.1.exe
Linux命令行下载:
# 手动安装
sudo yum install blas-devel lapack-devel
wget https://sourceforge.net/projects/mcmc-jags/files/JAGS/4.x/Source/JAGS-4.3.0.tar.gz
tar xf JAGS-4.3.0.tar.gz
cd JAGS-4.3.0
./configure --libdir=/usr/local/lib64
make -j 20 && make install或者conda安装:
conda install -c conda-forge jags r-rjags之后再安装infercnv,这个可以直接从BioManager 安装,或者git直接安装:
BiocManager::install("infercnv",force = TRUE)
##or
devtools::install_github("broadinstitute/inferCNV")数据读取
inferCNV requires: a raw counts matrix of single-cell RNA-Seq expression an annotations file which indicates which cells are tumor vs. normal. a gene/chromosome positions file
infercnv的输入数据有三个:
Raw Counts Matrix for Genes x Cells
细胞的注释文件:共两列,一列CB,一列细胞来源,tab分割,无列名;如果肿瘤细胞的注释类似malignant_{patient},则在后续聚类画图时,设置cluster_by_groups=T会根据patient的来源进行区分 该文件的CB可以小于count矩阵,此时inferCNV只会对这部分细胞进行分析;
基因排序文件:tab分割,无列名 只有该文件和count矩阵共有的基因才会被分析(可以去掉不想画图的基因,比如线粒体基因),且该文件的染色体顺序决定了最终热图的染色体顺序(有些文章的图,inferCNV热图的染色体顺序是颠倒的,问题就出在这儿)。我一般是根据cellranger提供的参考基因组gtf注释文件,得到基因排序文件,格式如下
WASH7P chr1 14363 29806
LINC00115 chr1 761586 762902
NOC2L chr1 879584 894689
MIR200A chr1 1103243 1103332实例操作
我们利用infercnv软件里面自带的例子做分析。主要步骤只有两步,如下
创建infercnv的对象
library(infercnv)
# Create the InferCNV Object
infercnv_obj = CreateInfercnvObject(raw_counts_matrix = system.file("extdata", "oligodendroglioma_expression_downsampled.counts.matrix.gz",
package = "infercnv"), annotations_file = system.file("extdata", "oligodendroglioma_annotations_downsampled.txt",
package = "infercnv"), delim = "\t", gene_order_file = system.file("extdata",
"gencode_downsampled.EXAMPLE_ONLY_DONT_REUSE.txt", package = "infercnv"), ref_group_names = c("Microglia/Macrophage",
"Oligodendrocytes (non-malignant)"))
## INFO [2022-12-03 20:28:38] Parsing matrix: D:/R-4.2.1/library/infercnv/extdata/oligodendroglioma_expression_downsampled.counts.matrix.gz
## INFO [2022-12-03 20:28:40] Parsing gene order file: D:/R-4.2.1/library/infercnv/extdata/gencode_downsampled.EXAMPLE_ONLY_DONT_REUSE.txt
## INFO [2022-12-03 20:28:40] Parsing cell annotations file: D:/R-4.2.1/library/infercnv/extdata/oligodendroglioma_annotations_downsampled.txt
## INFO [2022-12-03 20:28:40] ::order_reduce:Start.
## INFO [2022-12-03 20:28:40] .order_reduce(): expr and order match.
## INFO [2022-12-03 20:28:40] ::process_data:order_reduce:Reduction from positional data, new dimensions (r,c) = 10338,184 Total=18322440.6799817 Min=0 Max=34215.
## INFO [2022-12-03 20:28:40] num genes removed taking into account provided gene ordering list: 399 = 3.8595473012188% removed.
## INFO [2022-12-03 20:28:40] -filtering out cells < 100 or > Inf, removing 0 % of cells
## WARN [2022-12-03 20:28:40] Please use "options(scipen = 100)" before running infercnv if you are using the analysis_mode="subclusters" option or you may encounter an error while the hclust is being generated.
## INFO [2022-12-03 20:28:40] validating infercnv_obj去噪
关于cutoff参数,官网是这样说的:cutoff=1 works well for Smart-seq2, and cutoff=0.1 works well for 10x Genomics,表示基因在所有参照细胞中,表达count的平均值的最小阈值,10X数据更稀疏,所以这个值小 cluster_by_groups:先区分细胞来源,再做层次聚类 out_dir表示输出文件夹的名字,没有会自动创建。
创建 infercnv_obj 对象后,你可以通过内置的 'infercnv::run()' 方法运行标准的infercnv分析,如下所示:
infercnv_obj = infercnv::run(infercnv_obj,
cutoff=1, # cutoff=1 works well for Smart-seq2, and cutoff=0.1 works well for 10x Genomics
out_dir=tempfile(),
cluster_by_groups=TRUE,
denoise=TRUE,
HMM=TRUE)结果可视化
infercnv包也包含了画图函数plot_cnv:
library(RColorBrewer)
infercnv::plot_cnv(infercnv_obj, #上两步得到的infercnv对象
plot_chr_scale = T, #画染色体全长,默认只画出(分析用到的)基因
output_filename = "better_plot",output_format = "pdf", #保存为pdf文件
custom_color_pal = color.palette(c("#8DD3C7","white","#BC80BD"), c(2, 2))) #改颜色
我们这期主要介绍从肿瘤单细胞RNA-Seq数据中推断拷贝数变化。目前单细胞测序的费用也在降低,单细胞系列可算是目前的测序神器,有这方面需求的老师,联系桓峰基因,提供最高端的科研服务!
桓峰基因,铸造成功的您!
未来桓峰基因公众号将不间断的推出单细胞系列生信分析教程,
敬请期待!!
有想进生信交流群的老师可以扫最后一个二维码加微信,备注“单位+姓名+目的”,有些想发广告的就免打扰吧,还得费力气把你踢出去!

References:
Anoop P. Patel et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20: 1396-1401
Tirosh I et al.Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016 Apr 8;352(6282):189-96
Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016 Nov 10;539(7628):309-313. PubMed PMID: 27806376; PubMed Central PMCID: PMC5465819.
Venteicher AS, Tirosh I, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017 Mar 31;355(6332).PubMed PMID: 28360267; PubMed Central PMCID: PMC5519096.
Puram SV, Tirosh I, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017 Dec 14;171(7):1611-1624.e24. PubMed PMID: 29198524; PubMed Central PMCID: PMC5878932.