RNA分析流程

要理解整个流程,个人觉得可以按数据的四个流程来拆分:高通量测序,准备工作,上游分析,下游分析
【什么是高通量转录组测序】
所谓高通量测序技术是什么?
顾名思义,就是通量很高(对比sanger测序)的测序,一次性可以获得海量的数据,所以叫高通量测序。
转录组是什么?
转录组,一般指的就是某一时空条件下细胞所产生的所有转录产物,说人话就是,你的样品经过了某种处理,然后拿去提了总RNA,这个总RNA就是一个转录组。
理解高通量转录组测序的关键在哪?
首先是建库,我们建的文库用的是什么,rna吗?不是,那用的是什么?
cDNA,即rna拿去逆转录的产物,为什么要用DNA而不是RNA?
除了单链RNA不稳定外,还有一小部分原因是DNA的建库流程已经确定了,只要把RNA变成DNA后面流程完全一样,可以偷个懒,不过为了节约时间可以在一二链合成的时候直接加好接头,后面就连接头都不用加了,缩短建库的时间,一天可以轻松完成建库
其次就是什么是桥式pcr?
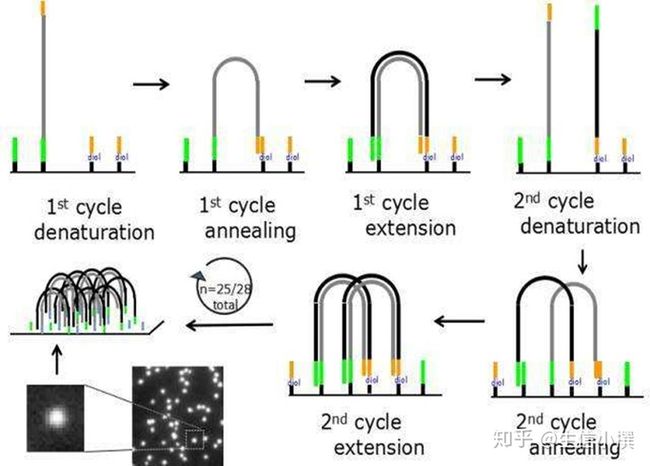
上面就是桥式pcr的流程,简而言之就是序列接头(adapter)一端被固定,然后另一端跟反应槽里的互补序列配对,呈现桥状,然后再进行pcr,故而称桥式pcr。
经过n轮桥式pcr之后,一个序列可以扩增到一个叹为观止的水平,故而通量就非常高了~
最后是测序信号是怎么得到的?
荧光基团,在所有的碱基上我都接了荧光基团

想更直观理解的这里放个illumina的官方介绍
illumina测序原理简介_哔哩哔哩 (゜-゜)つロ 干杯~-bilibiliwww.bilibili.com/video/BV1ht411q7Wh?from=search&seid=17159547191666876555正在上传…重新上传取消
【准备工作】
首先,我们拿到的原始序列文件就是fastq,那么怎么去理解fastq文件呢?
ST-E00126:128:HJFLHCCXX:2:1101:7405:1133
TTGCAAAAAATTTCTCTCATTCTGTAGGTTGCCTGTTCACTCTGATGATAGTTTGTTTTGG
+
FFKKKFKKFKF上面这里是一个fastq文件的格式,每一行代表什么呢?
第一行就是测序的坐标信息,即告诉你这条reads的名字是什么
第二列就是我们测到的序列
第三列就一个加号,没卵用
第四列,质量信息,对应着上面各个碱基,测得有多臭,具体多臭下面说怎么直观的看
需要了解的就这么多,如果要仔细了解,看下面这个帖子
孟浩巍:20160406 FASTA 与 FASTQ格式详解227 赞同 · 39 评论文章正在上传…重新上传取消
怎么对测序质量这些东西进行直观化?
有个东西叫做fastqc的软件,可以对fq文件进行质检,具体怎么看呢?看这个贴子
孟浩巍:20160410 测序分析——使用 FastQC 做质控327 赞同 · 78 评论文章正在上传…重新上传取消
在明白了自己的测序数据有多臭之后,我们就要将数据中低质量的部分全部剔除掉,剔除的软件有很多,类似Trimmomatic,fastp,cutadapter
一般给定的标准就是清除存在的所有接头序列,过滤掉q小于20的碱基,去除N碱基大于5%的序列,去除A与T或者C与G含量相差10%的序列,去除切除碱基后过短的序列,这个标准一般通用,具体可以根据自己数据去筛选。
想详细了解的看这个贴子
孟浩巍:20160420-序列比对前的准备工作92 赞同 · 42 评论文章正在上传…重新上传取消
准备工作完成之后,我们就得到了一份高质量的原始数据(clean data),从而正式进入分析工作
【上游分析】
无论是以前的bowtie2+samtools+cufflinks+deseq2,还是现在转录组的当红炸子鸡流程hisat2+stringtie+ballgown,其本质的工作流程其实是一样的,只不过使用的算法不同而已。
第一步叫做回帖,这一步是干嘛的呢?
首先,我们的fastq文件存储的数据是一个零散的状态,那要怎么样把它恢复到打断前的状态?
这里我们就需要一个模板,按照模板,我们把序列排序,大概就长这个样子
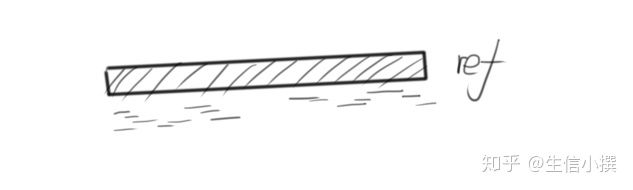
这里的ref就是模板,即参考基因组,而我们的fastq文件本质就是一条一条的小序列,在模板的指引下,我们得到了他们原本在基因组上应该在位置,这一步就是回帖的含义。
也即是bowtie2跟hisat2所干的事。
而关于回帖的细节,可以看这两篇
孟浩巍:踏踏实实做技术:BWA,Bowtie,Bowtie2的比对算法推导109 赞同 · 13 评论文章正在上传…重新上传取消
生信小撰:【生信常识】二代测序的比对算法浅析72 赞同 · 16 评论文章正在上传…重新上传取消
回帖完之后,我们的回帖信息会被输入到一个文本文件:SAM文件(二进制位bam文件)

sam文件有个头文件,即你看到这张图前面那样,存储着染色体的信息,还有你之前比对的指令,但这些不是我们需要了解的重点,我们需要来看看下面存储着什么?

第一列是什么?就刚刚fastq文件的第一列,就是这条reads的名字
第二列是什么?flag?太复杂了,不记了
第三列?染色体
第四列?染色体的起始位置
第五列?回帖的可信度,即回帖质量
第六列?第七列?看不懂,不管了
后面还有回帖上的序列
总的来看,所谓的sam/bam文件就是记录回帖的序列是什么,回帖上多少,回帖的质量行不行,回帖到什么位置。
而后就是用cufflinks或者stringtie结合注释文件gff/gtf,将转录本构建出来。
那么gff/gtf是什么?简而言之,gff就是记录了这个物种在哪个位置有功能,是gene还是调控因子。
孟浩巍:生物信息学100个基础问题 —— 第24题 GFF,GTF到底是什么?64 赞同 · 3 评论文章正在上传…重新上传取消
而cufflinks要做的事情就是将bam文件的比对信息跟gff的信息结合起来,拼出一条转录本
bowtie2做的事情是
而cufflinks做的事情则是这个
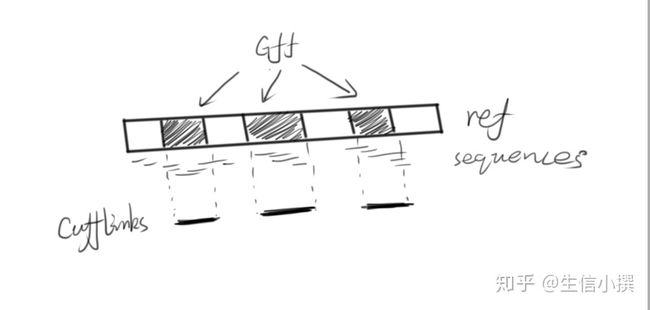
【下游分析】
当使用cufflinks构建得到raw count之后,我们就想比较不同处理间的差异在哪,那么这个时候我们可以直接比较吗?
当看到我这么问的时候,肯定就是说不可以。
那么,为什么不可以?
拿孟孟之前举的例子
问题1: 比如我有gene3,有1000条测序reads,gene4有2000条测序reads,那么我能否说gene4就一定比gene3的表达量高?
问题2: 比如我有gene1,有1000条测序reads,我的另一个处理条件下gene2有2000条测序reads,我能否就说geneA在处理条件下表达量降低了?
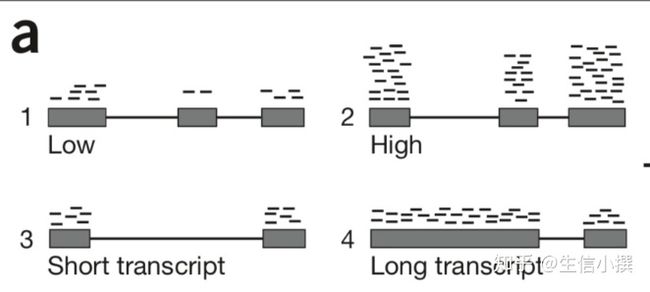
图1 ( Manuel Garber et al., Nature Methods, 2011 )
很明显,第一个问题,如果两个基因的长度不一致,那是无法直接比较的;而第二个问题,我们就需要考虑如何矫正了,而这个矫正值就是所谓的RPKM/FPKM/TPM,关于这些是什么
请看这个贴子
孟浩巍:生物信息学100个基础问题 —— 第35题 RNA-Seq 数据的定量之RPKM和FPKM93 赞同 · 13 评论文章正在上传…重新上传取消
孟浩巍:生物信息学100个基础问题 —— 第36题 RNA-Seq 数据的定量基本假设以及TPM35 赞同 · 17 评论文章正在上传…重新上传取消
当我们将所有的东西放同一个标准下,就可以进行比较了,而比较的时候,即肯定存在两个组才能进行比较,也就是我们的control跟treatment
以control为标准,比较treatment,我们就知道了差异究竟在哪些基因,即所谓的差异表达

现实计算肯定要复杂的多,但作为粗略理解,这样会比较容易理解
而当我们得到一堆差异基因之后,就通常要做所谓的富集分析,常见的有GO,KEGG。
以常见基于ORA算法的富集举例
本质其实就是一个超几何分布,常见的就是Fisher extract test
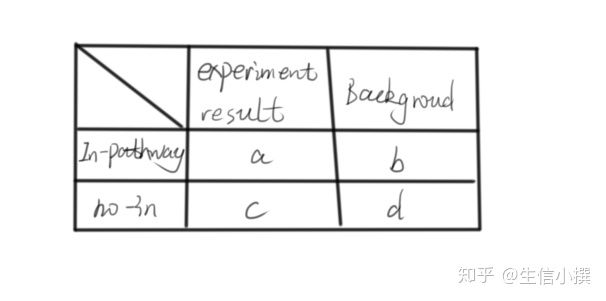
算出一个p值即可,然后自选标准,大于多少认为是显著的,认为某某通路上存在差异表达。
想更具体了解,可以看看这个视频
【GCModeller教程】基因组功能富集计算原理_哔哩哔哩 (゜-゜)つロ 干杯~-bilibiliwww.bilibili.com/video/BV1R4411d7xe正在上传…重新上传取消
本期内容就到这里,还望各路大神轻喷,同时欢迎各位大神指点一下哪里可以写得很通俗而不失谨慎,方便新入门的小伙伴更好的理解整个分析流程~