Pymol入门教程--基础
Pymol入门教程–基础
软件界面介绍
内置demo介绍
打开PyMOL,点击1.1菜单窗口的Wizard菜单,然后点击Demo->Representations然后在2.3对象窗口和2.4模式窗口之间会出现各种示例:
- Representations
- Cartoon Ribbons
- Roving Detail
- Roving Density
- Transparency
- Ray Tracing
- Sculpting
- Scripted Animation
- Electrostatics
- CGOs
- Molscript/R3D Input
- End Demonstration 关闭示例

设置和查看工作路径
点击菜单窗口File->Working Directory->Change可以查看现在的工作目录在哪里,也可以设置新的路径作为工作目录。
工作目录的作用:默认文件的打开和保存都是从该文件开始。下载文件的保存位置也是在工作目录。这一点与R语言类似,我也是习惯一个项目新建一个project,方便以后查看。
工作目录设置习惯
建议:
- 不同项目设置不同的工作目录
- 设置一个默认工作目录
- 不要把工作目录设置在软件安装目录
双击PDB文件打开PyMOL,会自动切换工作目录到该PDB文件所在目录。
从软件安装处打开PyMOL,则工作目录为软件安装目录。
下载蛋白
从PDB网站上下载蛋白,如4hbk.pdb.也可以直接通过PyMOL下载蛋白,点击菜单栏中的File->Get PDB.如下图图所示,在PDBID对应的框中填入PDB的编号就可以了,不要包含后缀.pdb。

点击Download,你会发现PyMOL会自动加载该蛋白,在工作路径D\PyMOLstartedManual下面出现4hbk.cif文件,随着PDB解析的结构越来越大,PDB格式文件的局限性就暴露出来,不能超过9999个原子,因此正在逐渐用cif 格式取代pdb格式。
介绍"ASHLC"
蛋白的展现形式(show)
在2.3对象列表窗口中,我们可以看到现在有2个object名字,
- 1个是all,all 不是真实的object,它代表了所有的object
- 1个是4hbk,4hbk就是我们刚刚载入的蛋白每个object 都有对应的ASHLC操作,如下图所示:

- Action 主要包含了对object的常用操作的集合,如复制、删除object对object加氢,展示Object等:
- Show 将object 渲染成cartoon、line、stick lines sphere surface mesh dots ribbon 等模式:
- Hide 根据object的状态或者描述进行相应的掩藏;
- Label显示object中残基原子等名称或者属性-Color对Object进行着色。
下面对Show 着重讲解:show有2类操作方法:
- show as分别点击S->as->cartoon和S->as->stick,
我们可以观察到AS模式是把原有的渲染模式抹除后再重新渲染,经过上述操作后仅仅显示stick形式 - show点击S->as->cartoon 再点击S->stick;
我们可以观察到SHOW方法,是保留原有的渲染,再添加新的渲染。
我们先对4hbk object点击S->as->cartoon.然后点击S->as->stick,效果如图Fig5
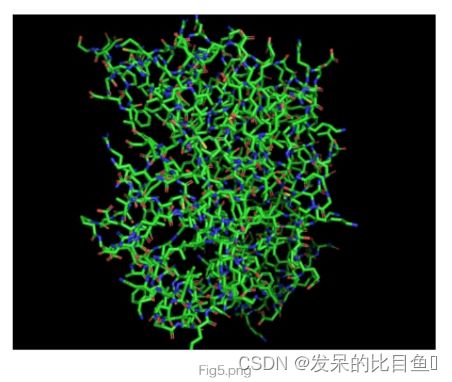
我们先对4hbk object点击S->as->cartoon.然后点击S->stick.效果如图Fig6

蛋白对象的简单平移、旋转、缩放
首先将鼠标移动到2.1可视化窗口 - 平移,按住鼠标中键不放,然后上下左右移动,进行体会,蛋白会随着鼠标而移动
- 旋转,按住鼠标左键不放,然后上下左右移动鼠标,蛋白会进行旋转
- 缩放,按住鼠标右键不放,然后上下移动,蛋白会进行缩放
- 切割滚动鼠标中键,建议将蛋白渲染成surface模式,然后滚动鼠标中键当软件不能正常使用上述操作,可以点击
File->Reinitialize->Original Settings(推荐)
File->Reinitialize->Everything 注意的是该操作会删除当前所有的Object.
Action
第一部分:常用显示操作
点击A->preset->simple显示蛋白的简单形式
点击A->preset->ball and stick 显示球棍模型
点击A->preset->b-factor putty 基于bfactor数值显示蛋白的柔性
点击A->preset->publication 高质量出版标准
第二部分对象的操作
删除水分子A->remove waters
该操作属于红色警告操作,不可逆操作。删除水分子后,无法通过Ctrl-Z进行撤销。
增加删除氢原子
在line和stick模式下面可以看到H原子,cartoon模式下面看不到氢原子,因此在line或者stick模式下,执行下述操作。
A->Hydrogens->remove 删除所有氢原子
A->Hydrogens->add 增加所有氢原子
A->Hydrogens->remove non polar 删除所有非极性氢原子,我们可以看到C上的氢原子全部被删除
A->Hydrogens->remove再次删除所有氢原子
A->Hydrogens->add polar 增加极性氢原子
第三部分对象的复制剪切删除重命名
复制A->Copy to object->new
我们会得到一个名为new的object.为了和4hbk object进行区分。
对4hbk 的obect 点击C->cyan 和S->AS->cartoon
对obj01点击C->green和S->as->stick
重命名obj01->4hbk_02对obj01
点击A->rename object
删除A->delete object
如果要删除当前所有的object的话,可以在1.3命令输入窗口输入delete ll。
剪切适合选中的对象A->Extract
我们可以通过剪切的操作把配体和蛋白分开,首先选中配体,然后点击A->extract object 就可以了,原来结构中的配体就跑到obj01中了,这样就分开了蛋白和配体。下面会有具体的例子来说明。
第四部分Action->generate操作
- 显示蛋白的静电势图Action->generate->vacuum_electrostatics->protein contact potential 就可以了。
查看小分子和蛋白的氢键作用
由于4hbk蛋白中没有小分子,这里我以PDB id:为例演示。点击A->preset->ligand sites效果如图所示,其中黄色的虚线就是氢键。
对标注的氢键,查看距离和角度,进一步确定氢键的合理性和强度。
查看小分子和蛋白的相互作用,推荐软件 schrodinger中ligand interaction。制作相互作用的二维图,根据提示,然后再在pymo确认并绘制这些相互作用。
蛋白对象的Hide操作
和remove delete操作相比,hide操作更加温和,把不需要的东西暂时掩藏起来,通过Show可以重现显示出来。我们先上述方法,构建4hbk和4hbk_02两个object,这一次把4hbk设置成cyan颜色的cartoon,4hbk 02设置成green颜色的ribbon,如图所示。如果要掩藏4hbk_02.有2种方法-对4hbk_02点击H->ribbOn-直接点击4hbk_02的名字。当然,这里展示的时候要选择不同的展示方式,如果是相同的展示方式,是显示不出来的。

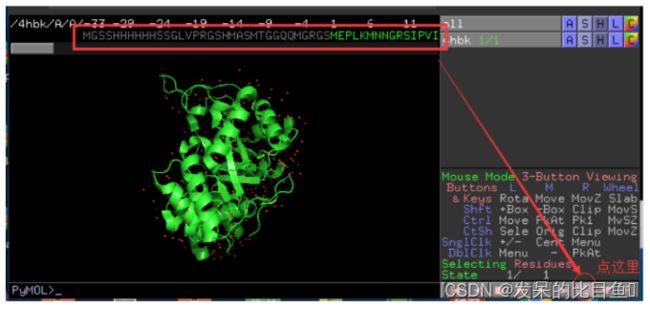
查看该结构的序列(Sequence)
点击左下角的S,即可显示蛋白的序列信息,如图所示,再点击一次S,则掩藏序列信息。S是单词Sequece的缩写。
应用:选择蛋白的特定残基
我们通过软件D3Pockets可以确定4hbk中口袋中氨基酸残基的组成:
ILE15-TRP20,TYR39,ASP43-ALA45,VAL47,TYR48,LYS77,TRP79,ASN80,LEU108,HIS110,TRP111,LEU115,PHE122,LEU130,SER159,ASN160,GLN181,GLU183,HIS185,185207 208 209210211222223 240241243244255256257258263264266267270292293294295298306
我们点击Sequence,并将Selecting模式切换为residues.然后基于残基编号进行选择。
把上述残基选中以后会临时保存在sele的object中,我们把它复制到obj01的Object中,
并重名为Pocket.然后对Pocket的Object,显示其表面,结果如图所示。

蛋白对象的着色操作
pymo中内置了多种不同的着色方案,如图所示:
按照原子类型着色
点击object上的C按钮>by element按照二级结构着色
按照b-factor着色
按照整体着色设置背景颜色
背景颜色
默认是黑色的,发表文章的时候背景颜色通常设置为白色。
点击菜单栏中的Display->background->white
制作b-factor-value图
点击A->preset->b-factor putty.
对象的Label操作
添加label
选择需要Label的对象|然后点击对象上的L按钮,根据需要.可以标记残基的名字,原子的名字.范德华半径、元素的名字等。
- L->residue在a碳原子上标记其残基名字和编号(常用)
- L->residue name 在所有原子上标记残基名字(不常用)
- L->clear 删除该对象上所有的Label
- L->element symbol显示对象上所有原子的元素名字
- L->vdw radius产看原子的范德华半径
对于蛋白醛糖还原酶(PDB ID:4HBK),我们在UniProt 数据库中查询得到,其口袋中的重要残基有:TYR48、LYS77和HIS110。
我们对这三个残基展示其为stick模式,并掩藏主链,并通过label按钮标注其氨基酸残基的名字。移动Label,使其更加清晰可见。具体操作流程如下:
6. 载入4hbk 蛋白load 4hbk.cif
7. 在4hbk object 上点击S->as cartoon
8. 点击右下角的sequence开关按钮,选择TYR48LYS77HIS110三个残基,得到sele 临时object
9. 在sele object 上点击 S->stick,H->main chain
10. 在sele object 上点击L->residue
11. 设置背景为白色,点击菜单栏中Display->background->white
12. 点击Mouse Mode 切换为editing模式
13. 按住ctrl键不放,然后将鼠标移动到残基标签上方,并按下鼠标左键不放,然后移动鼠标,就可以调整标签的位置。
14. 调整结束后,点击Mouse Mode 切换为view 模式

移动标签label
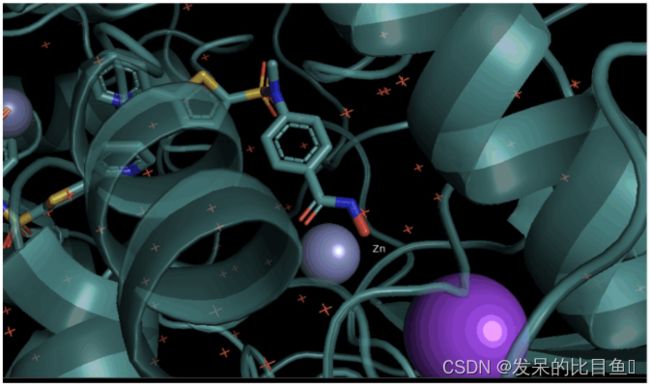
以PDB ID:1w22蛋白为例,为其中的锌离子添加标签。
sphere模式下不能添加label;借用PS中复制图层的概念。为锌离子添加label。
step1 选中锌离子;
step2 复制锌离子;仅仅显示该object,并显示为lincore模式
step3 添加label(右击new-pseudoatom-label):进入edit模式,按住ctrl移动label,完成后切换回viewing模式
step4 显示所有的object.
设置label的样式
调整配体的位置
pymol中包含配体和蛋白,那我们如何调整配体和蛋白的相对位置。
以PDB ID:1hkv为例,其中配体小分子为PLP。
step1 打开蛋白文件,定位到想要移动的配体。
step2 extract 操作,提取配体为新的object.并进行重命名。
step3 对蛋白进行固定(最大化窗口),
step4 在edit模式下,按住shit 就可以通过鼠标移动配体。(只是空间位置的变动,center还是不变的)

测量距离
点击菜单栏中的Wizard->Measurement 就可以进行距离测量。然后分别选择两个2个原子就可以显示这2个原子的距离。这里蓝色表示N端,红色表示O端。
比如我们想测定TYR48中侧链上的O原子到Lys77侧链N原子的距离。如下操作即可:
- 点击Wizard->Measurement;打开了Measurement 面板,位于object list面板下方;
- 选好残基Y48和K77,鼠标点击TYR48侧链上的O原子和LYS77侧链上面的N原子;这里为了方便,我把cartoon部分和主链部分给隐掉
- 如要测定其他2个原子的距离,鼠标继续点击选择2个原子就可以了;
- 测定完成后,点击Measurement 面板中的done就可以了。

突变氨基酸残基
如把4hbk中的arg-40突变成lys-40.
在菜单中点击wizard->mutagenesis->protein
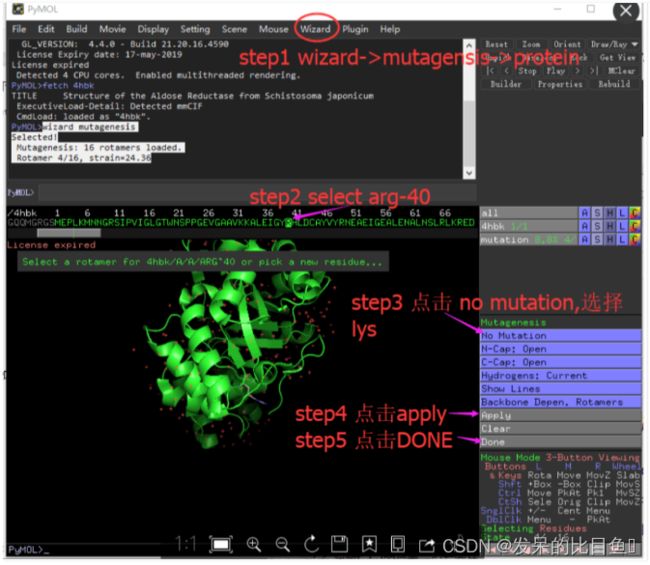
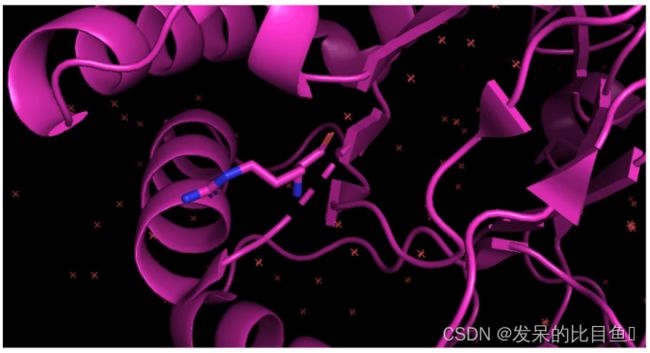

其他技巧
设置透明度
鼠标操作只能基于显示模式进行设置,如:cartoon surface sphere stick等
- 将cartoon设置成透明的,透明度50%;点击莱单栏中的Setting->Transparency->cartoon->50%;
- 将Surface 设置成透明的,透明度50%;点击菜单栏中的Setting->Transparency->surface->50%;
- 将Stick设置成透明的,透明度50%;点击菜单栏中的Setting->Transparency->stick->50%;
- 将sphere 设置成透明的,透明度50%;点击菜单栏中的Setting->Transparency->sphere->50%;除了可以设置透明度外,还可以设置透明的方式,有如下几种透明模式:
- Uni-layer;点击菜单栏中的Setting->Transparency->Uni-layer
- Multi-Layer;点击菜单栏中的Setting->Transparency->Multi-Layer
- Multi-Layer(real time oit):点击菜单栏中的Setting->Transparency->Multi-layer(real time oit)
- Fast-ugly;点击菜单栏中的Setting->Transparency->Fast-ugly在Label操作中,我们将整个蛋白展示为cartoon,并将HIS等3个残基展示为stick模式;这时候我们设置cartoon为透明50%,用4种不同的透明模式渲染,效果如下:
雾化(fogging)处理
PyMOL中支持各种雾化处理,保证depth_cue是开启的。
前面清晰、后面雾化可通过滚动鼠标中键进行调节。向上滚动,雾化程度减轻;向上滚动,雾化程度加深。
完全不想要雾化处理,可以点击 Display->Depth cue(Fogging)取消。
或者使用命令关闭。
设置不同的光照模式
PyMOL2中内置5种不同的光照模式:default,metal(金属),plastic(塑料),rubber(橡胶),X-ray。
点击Plugin->lighting Settings进行设置不同的光照,如下图所示图。
选择模式
根据不同的需求,切换到不同的选择模式,快速选择自己想要的原子;
- Residues残基选择模式
- Atoms原子选择模式
- Molecules分子选择模式
- chain 链选择模式
- Objects 模式
- C-alphasa碳原子模式
- Segments片段模式
我们先将蛋白Show->as wire;然后把MET-1第一个氨基酸残基show stick:如图所示,金色的表示S原子。
从上图我们也可以看到,不同模式的区别。这里我简单解释下:
res模式:我们点击的是硫原子,硫原子所在的res是MET-1,因此选中的是MET-1
chain模式:我们点击的是硫原子,硫原子所在的chain A.因此选中的是所有属于chainA的原子。
mol模式:我们点击的是硫原子,pymol中是通过共价键来区分是否属于一个分子,4hbk蛋白中间缺失了部分残基,整个蛋白分成了2部分。
这里选择的是硫原子所在的那一部分。
保存或者导出结果
PyMOL和PhotoShop 有点类似,PyMOL中的Object类似于PhotoShop中的图层。
- PyMOL可以将会话保存为pse文件,File->Save Session;pse文件类似于PS中的psd文件,方便修改调整。
- PyMOL可以将object 导出为结构文件,File->export molecule.然后从selection的下拉框中选择需要导出的object.all代表所有的object;enable代表的可见的object;
- 保存图片,File->export image as->png;在新版本的,可以使用右上角的Ray/Trace按钮,设置图片大小分辨率,进行保存图片。
- 保存动画,File->export movie as->mpeg;PyMOL仅仅内置了mpeg_encode编码视频方式,默认只能保存mpg格式的动画。可自行下载ffmpeg.从而可以保存为多种格式,如gif.mov.mpg等
设置pse的版本号,建议版本为0.99,这样PyMOL和PyMOL2都可以打开pse文件
删除蛋白的一条链,然后保存为pdb
step1. 切换到chain模式
step2. 点击需要需要删除的Chain就可以选中chain
step3. 对新产生的sele object进行删除。
移动object在列表中的顺序
鼠标右击待移动的object.按住鼠标不放,移动鼠标,调整object在列表中顺序。
参考
https://wenku.baidu.com/view/628f6d6ccc84b9d528ea81c758f5f61fb73628ee.html