【生物信息】ecDNA染色体外环状DNA的识别——AmpliconArchitect算法
ecDNA染色体外环状DNA的识别
- 写在前面
-
- 实验环境
- 原理简介
-
- 算法流程
- 实现细节
-
- 区间搜索
- 区间重排,分区和可视化
- 断点图的构造
- 环分解
- 实验步骤
-
- 数据的获取
- 质控分析
-
- conda的安装
- AA算法的准备
-
- 下载参考基因组
- bwa工具
- 安装samtools
- CNVkit
- 使用PAA
- AA算法
-
- 安装AA算法
- 输入
- 使用方法
- 成图输出
写在前面
这个项目是我大三时期(2020年)做的SRTP,是与我们学校的生物信息专业的同学一起做的。生物上的东西我不是很了解,我只是帮我的同学找了一些工具并使用这些工具完成我们的SRTP。
SRTP题目是:染色体外DNA的识别与分析
我们主要是参考了这篇2019年发表在《nature》上的论文:Circular ecDNA promotes accessible chromatin and high oncogene expression
他们论文中提到了他们使用的AA算法,并提供了AA算法项目的开源地址,有了AA算法,我们就可以通过基因序列找到环状DNA,并把这些基因序列组合成环,输出成图片。
以下是我们在某个癌症细胞基因序列上提取到的ecDNA扩增子的重建结果:
实验环境
操作系统:deepin15.5 sp2
python: python3.8
原理简介
算法流程
AA采取许多步骤来预测扩增子(amplicons)的结构:
step1:间隔集确定:确定要重建的每个扩增子的间隔列表。
step2:SV检测:利用每个扩增子中的覆盖率和不一致的读段双簇(coverage and discordant read pairs)来检测拷贝数变化和结构变异。
step3:断点图构造:构造一个断点图,该断点图由序列egdes(基因组片段),断点边(连接的因组位置对)和源顶点(可选)组成,并预测所有边的拷贝数。
step4:循环分解:将断点图分解为简单的循环,以简单的方式表示预测的扩增子(amplicons)结构。
step5:交互式循环合并:提供一个界面,以交互方式合并或修改环视图(cycles)以探索候选结构。
实现细节
AmpliconArchitect实施了一系列步骤,从种子区间开始,最终重建了扩增子的完整结构,并为每个阶段提供了有益的结果:
区间搜索
在此步骤中,AmpliconArchitect从种子区间开始,迭代地标识属于扩增子的区间列表。首先创建一个存储种子区间的最大堆数据结构。
AmpliconArchitect重复以下步骤,直到最大堆为空或经过10次迭代。
在每次迭代中,AmpliconArchitect选择一个区间并以拷贝数敏感的方式确定不一致成对读段双簇,并选择双簇,其配偶读段比对位置位于先前看到的区间之外。然后,通过查询扩展部分是否被扩增,尝试扩展双簇。如果查询段的窗口(10kbp)至少有20%且覆盖深度>,或者小于20kbp且包含至少两个不一致边(双簇大小对应于拷贝数=2),则分类为扩增。然后,AmpliconArchitect迭代地将扩展部分的大小加倍,直到发现该扩展被扩增,然后通过迭代将扩展查询的大小减小一半,来有效地扩展查询的两部分。如果AmpliconArchitect能够成功扩展查询的双簇,则它将其进一步扩展100kbp,并记录扩展区间以用于将来的迭代。在AmpliconArchitect记录了所有放大的邻近区间之后,AmpliconArchitect标记所看到的区间,根据连接到先前看到的区间的不一致成对读段的数量更新了最大堆,并贪婪地选择堆顶部的区间以进行下一次迭代。AmpliconArchitect报告延伸步骤中所有扩增的区间。
区间重排,分区和可视化
AmpliconArchitect计算了所有覆盖范围的均值移位边界和相应段的初始拷贝数估计值。然后,它创建了不一致成对读段双簇列表,其双簇大小阈值由拷贝数估计值(或差异)确定。此外,对于没有找到匹配的不一致读段双簇的均值移位边界,它对双簇大小阈值仅为两个读段对的不一致成对读段执行敏感本地搜索。最终,它创建了所有重排的基因组位置集,并带有不一致成对读段双簇或均值移位边界(没有匹配的不一致成对读段)。使用这组位置,它将所有区间划分为序列边。对于第二阶段的输出,AmpliconArchitect创建了一个单独的图,称为结构变异视图,该图显示了带有覆盖深度直方图的区间集,均值移位段的初始拷贝数估计以及不一致成对读段双簇。
断点图的构造
AmpliconArchitect使用序列边分区构造了一个断点图。对于每个序列边,它创建了两个断点顶点,标记了基因组片段的起点和终点,并添加了连接两个顶点的序列边。AmpliconArchitect用特殊的源顶点扩展了顶点集。对于每个不一致成对读段双簇,如果两个簇都属于区间集,则会添加一个不一致断点边,该断点边连接相应序列边的各个端点。对于在区间集之外具有一个簇的所有双簇,它引入了一个源断点边,该边将源顶点连接到与扩增子区间内的簇相对应的断点顶点。它还添加了源断点边,这些源断点边将源顶点连接到与均值移位顶点相对应的断点顶点(没有对应的不一致双簇)和扩增子区间的端点。最后,AmpliconArchitect将对应于每个区间内连续序列边的断点对与断点边保持一致。对于每个序列边和断点边,AmpliconArchitect记录分别映射到该边的读段和读段对的数量。正确重建的断点图表示所有扩增子结构的叠加。每个环结构形成一个交替序列和断点边的环。端点与环外的基因组位置相连的线性结构可以表示为在源顶点处开始和结束的交替闭合步态。断点图的构建可能并不总是完整的,因为缺少边会导致结构的最终预测不准确。如果AmpliconArchitect无法检测到不一致断点边,但通过均值移位边界检测到重排的位置,则相应的扩增子结构将被表示为从源顶点开始和结束的一个或多个游走,从而部分缓解了此问题。此后,我们将断点图中的环定义限制为具有交替的序列边和断点边的闭合遍历,但该遍历可能包含最多两个连接到源的连续断点边。
使用平衡流优化的拷贝数估计:接下来,我们注意到每个边的拷贝数是每个扩增子结构的拷贝数的总和,其中边的每次遍历分别进行计数。结果,图中的拷贝数遵循平衡流动特性,其中序列边的拷贝数与连接到序列边的每个断点顶点的断点边的拷贝数的总和相匹配。AmpliconArchitect使用由拷贝数、边长度和序列覆盖深度参数确定的参数,将比对到每个边的读段片段的数量建模为Poisson分布。使用凸优化软件包Mosek获得平衡流的最佳解决方案。对于第三阶段的输出,AmpliconArchitect将图边及其拷贝计数报告为文本输出。
环分解
如上所述,线性或环扩增子结构可以在断点图中表示为一个或多个环。但是,即使使用正确的重构和对断点图的拷贝数分配,也无法始终明确地推断出环,尤其是在反复遍历边的情况下。相反,可能有两个或更多可能的环集和相关联的拷贝数,这样,将每个集合内的环组合在一起,最终可以得到具有相同拷贝数分配的相同断点图。在这里,环的组合仅意味着对每个环中每个图形边的拷贝数求和。枚举所有可能的扩增子结构并不总是可行的,因为可能的结构数目可能成倍增加。为了解决这个问题,首先观察到沿同一方向多次遍历边的环可以分为两个较小的环,相反,可以将两个环合并以形成原始环。但是,如果我们迭代地将多个环合并在一起,则更改环合并的顺序会产生不同的结果结构,这些结构都具有相同的边和拷贝数。基于此观察结果,AmpliconArchitect将断点图分解为简单的环,目的是使用相对较少的环表示大量的扩增子结构。我们将一个简单的环定义为一个环,该环在每个方向上最多遍历任何序列边一次,因此不能分为更小的环。我们将断点图的分解定义为一组具有拷贝数分配的简单环,以使所有简单环中任何边的拷贝数都等于断点图中的拷贝数。尽管断点图可能具有多个分解,并且单个分解中的简单环可能不会始终结合在一起以形成每种可能的扩增子结构,但这些情况要求断点图具有某些预期在小部分扩增子中出现的复杂模式。取而代之的是,AmpliconArchitect使用多项式时间试探法分解了断点图,该迭代法迭代地选择了具有最高拷贝数的简单环,并从断点图中的相应边减少了拷贝数。使用该算法,AmpliconArchitect可以优先考虑具有最高拷贝数和环的结构,这些结构出现在大量结构中,从而提供了一种突出扩增子重要特征的有意义方式。AmpliconArchitect提供了一个文本文件,其中包含每个简单环内读段的有序列表。此外,AmpliconArchitect提供了一个简单环的交互式可视化效果,称为环视图,可以将其合并为更大的环,以研究可能的扩增子结构。在环视图中,AmpliconArchitect在结构变异视图中显示了与其基因组位置比对的片段,并将连续的片段放置在连续的线上。如果两个环包含重叠的片段,则用户可以选择环,它们的重叠片段将这些环合并以形成更大的环。环视图提供了一种在可视化基因组位置和注释的同时解释环结构的方法。
实验步骤
数据的获取
在做实验时,首先就要解决的是数据问题。
NCBI(BioProject=PRJNA506071)Circular extrachromosomal DNA drives massive oncogene expression and chromatin remodeling]上可以下载到实验样本癌症基因的数据,具体操作如下:
选择GBM39HSR的WGS(全基因组鸟枪测法)数据:

点击SRR9157087,进入如下界面,选择Data access:
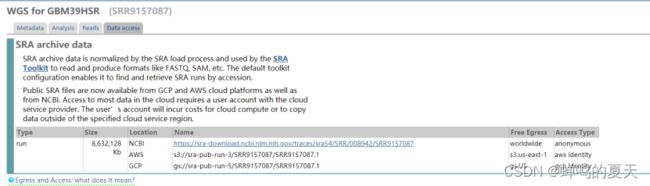
点击上图链接,便可以下载.run格式的数据,需要用到SRA Toolkit工具将其转换为fastq格式文件,
SRA Toolkit工具的下载地址:https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit
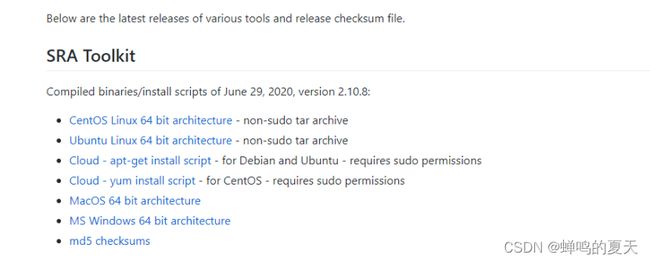
选择对应操作系统的版本。比如windows版本的安装以及使用指南:
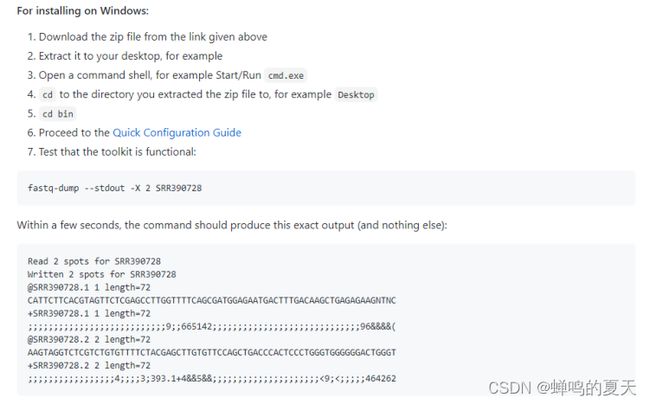 ①下载zip文件,解压缩
①下载zip文件,解压缩
②打开cmd,cd到“解压缩后的目录/bin”
③简单使用SRA Toolkit:
1)命令prefetch 可以从远程站点下载文件prefetch SRR1553610
2)将sra转换成fastq:fastq-dump SRR1553610
3)sra转换成fasta:fastq-dump --fasta 50 SRR1553610
4)将双端测序文件分开:fastq-dump --split-files SRR1553610
eg:使用命令“fastq-dump SRR9157087 -gzip”将8G多的数据转化为fastq格式数据的
质控分析
然后使用FastQC进行质控分析:
首先要安装FastQC,具体参考这篇博文
conda的安装
①安装conda
浏览器搜索 miniconda 清华
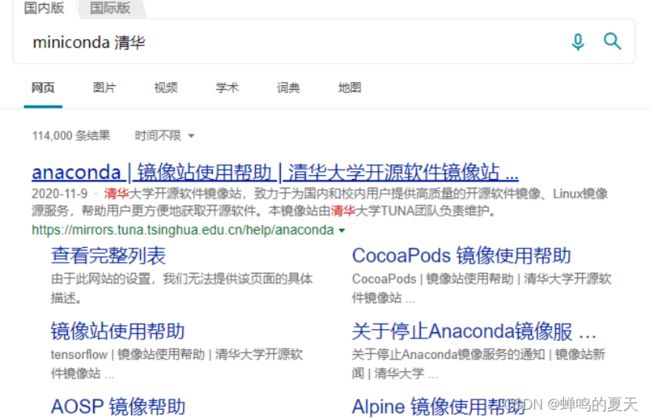
找到miniconda,点击链接,
将光标定位在 Miniconda3-latest-Linux-x86_64.sh(电脑环境是:deepin15.5 sp2),然后右键,选择复制链接地址,然后再在linux在使用以下命令下载miniconda
wget -c “链接地址”
安装刚刚下载的Miniconda,bash就是运行.sh文件的意思
bash Miniconda3-latest-Linux-x86_64.sh
然后根据提示按enter键或者输入yes,即可。
conda安装成功之后,再添加一下镜像,方便以后下载软件。逐行运行下面的命令,添加镜像:
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
conda config --set show_channel_urls yes
ref:https://www.jianshu.com/p/fab0068a32b4
可能还有别的安装方式,但是通过做这个SRTP我发现用conda真的方便太多,建议大家都用conda,这让我以后的实验也少走很多弯路。我第一次做时,我是在装samtools时才第一次接触到conda,因为不怎么会用所以感觉很麻烦,后来做类似的东西做多了才熟悉起来的,才发现真的太方便了。
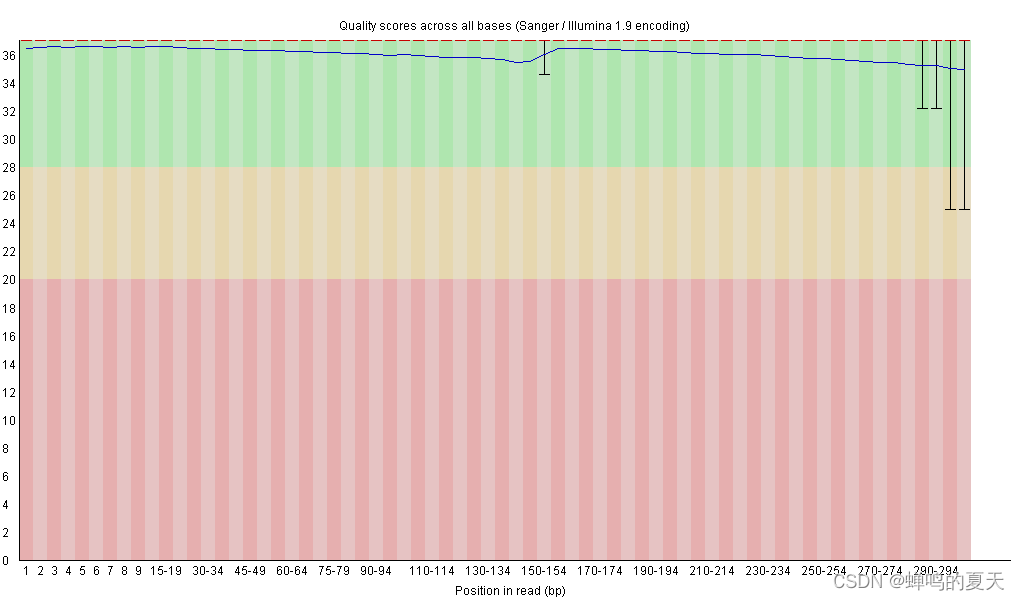
输入命令,得到了一个html,SSR9157087质控分析结果如下:

指导我们做这个SRTP的学长说质量不太行,叫我们换数据,于是我们换成了这个数据:WGS for GBM39
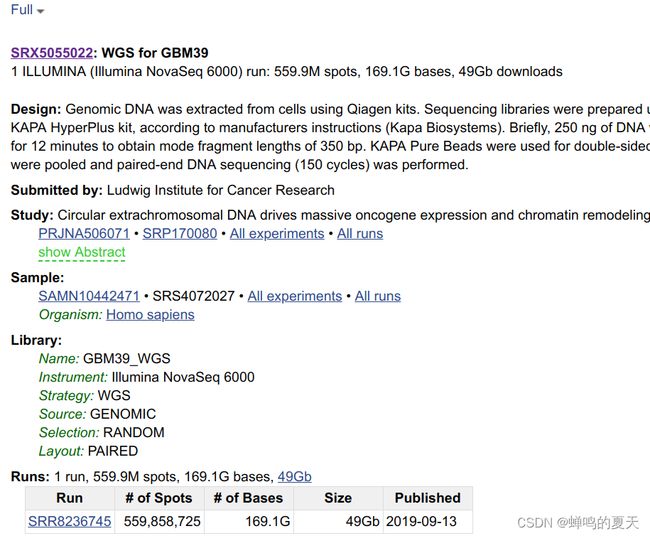
SRR8236745质控分析如下:
AA算法的准备
AA算法在直接调用前有一个快速入门的方法,使用准备AA准备我们需要的输入的参数。
PrepareAA 支持 hg19(或 GRCh37)、hg38 以及小鼠基因组 mm10(或 GRCm38)
根据PrepareAA的自述文件,我们需要解决以下问题:

下载参考基因组
比对需要用到参考基因组,以此来确定样本的可用性,本小组用到的参考数据组为人类基因hg38组,参考基因组fastq数据的下载方法如下:
到网站GENDODE下载人类基因数据,选择下图第5条,下载解压

bwa工具
到网站下载压缩包,解压,安装:
管理员权限下:
tar jxvf bwa-0.7.15.tar.bz2
cd bwa-0.7.15
make
BWA使用步骤:
①#加入临时环境变量
echo export PATH=”bwa所在目录”
②使用BWA构建参考基因组的index
bwa index [-p prefix] [-a algoType] .db.fasta>
-p 输出文件的前缀
-a 选择构建索引的算法
>is 是默认的算法,是由于其快速简单,但是不适合超过2G的基因组,因此人类基因组需要指定另一个算法
>bwtsw 对于基因组大于2G的,人类基因适合使用该算法
例子:
bwa index -p hg38 -a bwtsw GRCh38.p13.genoma.fa
③使用BWA-MEM算法进行比对
bwa mem [-aCHMpP] [-t nThreads] [-k minSeedLen] [-w bandWidth] [-d zDropoff] [-r seedSplitRatio] [-c maxOcc] [-A matchScore] [-B mmPenalty] [-O gapOpenPen] [-E gapExtPen] [-L clipPen] [-U unpairPen] [-R RGline] [-v verboseLevel] db.prefix reads.fq [mates.fq]
-t 设置线程数
-M 将较短的split hits标记为secondary,与picard兼容
例子:
bwa mem -t 5 -M GRCh38.p13.genome.fa SRR9157087.fastq > SRR9157087.sam
安装samtools
安装:
conda install samtools
成功后,测试,输入“samtools”将会有很多命令解释。

将sam格式文件转换为bam格式
samtools view -b SRR9157087.sam > SRR9157087.bam
CNVkit
CNVkit推荐使用python3,因此我们可以用conda创建一个虚拟环境。
conda create -n py38 python=3.8
激活环境
conda activate py38
后续的安装可以参考这篇博文。好像安装花了一下午,具体细节没有记录,遇到问题在网上搜一搜基本都有解决方法。
值得注意的是,CNVkit 需要 R 版本 3.5 或更高版本。这在许多 Linux 系统上不是标准的。如果需要,在后续使用时请指定–rscript_path /path/to/Rscript本地安装的当前 R 版本。
使用PAA
使用docker安装PAA,首先我要在我的电脑上安装docker(操作系统deepin15.5),可以参考这篇博文。
![]()
还需要License for Mosek,具体操作如下:
获取许可文件mosek.lic(https://www.mosek.com/products/academic-licenses/或https://www.mosek.com/try/)
export MOSEKLM_LICENSE_FILE=.lic> >> ~/.bashrc && source ~/.bashrc
下载 AA 数据存储库并设置环境变量 AA_DATA_REPO:
从此处下载 AA 数据存储库
设置环境变量 AA_DATA_REPO 指向 data_repo 目录:
tar zxf data_repo.tar.gz
echo export AA_DATA_REPO=$PWD/data_repo >> ~/.bashrc
source ~/.bashrc
使用方法如下图:

具体的参数详解请参阅项目自述文件。
AA算法
有了上面的磨练,安装和使用AmpliconArchitect就方便多了,readme里写得超级详细,在此就不作赘述。需要注意的是,它使用的python版本是2.7,因此必须注意python的版本,建议用conda建立正确的虚拟环境,版本高了会有不兼容的地方,报很多莫名其妙的错,具体实现的细节我不太记得了,如果遇到了报错可以先检查一下是不是版本问题。
安装AA算法
方法一:
像PAA一样,可以用docker安装
方法二:
github源代码
输入
BAM文件:WGS读取映射到人类基因组
BED文件:一组种子(seed)间隔,用于搜索和重建样品中的扩增子(amplicons)。用户应为每个扩增子(amplicons)提供至少1个种子。
使用方法
$AA --bam {input_bam} --bed {bed file} --out {prefix_of_output_files}
Github 源代码中提供了执行脚本$AA,具体路径取决于使用的安装选项:
码头工人形象: AA=AmpliconArchitect/docker/run_aa_docker.sh
Github 源码: AA=python2 AmpliconArchitect/src/AmpliconArchitect.py
具体的参数详解请参阅项目自述文件。
成图输出
输出了文件,你以为就成功了吗?其实没有,AA算法并没有得到一些优美的圆形,原来是因为还需要下游工具(CycleViz),它的用法与上面的工具使用方法大同小异,具体怎么用就请参看它们的自述文件吧。