iMeta | 南农沈其荣团队发布微生物网络分析和可视化R包ggClusterNet
点击蓝字 关注我们
ggClusterNet:包含多种基于模块可视化布局算法的微生物网络挖掘R包

https://doi.org/10.1002/imt2.32
SHORT COMMUNICATION
Volume1, Issue3

●2022年6月13日,南农沈其荣团队在iMeta在线发表了题为“ggClusterNet: An R package for microbiome network analysis and modularity-based multiple network layouts”的文章。
● 该文章开发了名为ggClusterNet的R包,展示微生物网络模块化信息,用于微生物网络数据挖掘和跨域网络数据挖掘。
● 第一作者:文涛,谢鹏昊
● 通讯作者:刘永鑫 ,袁军 ([email protected])
● 合作作者:杨胜蝶,牛国庆,刘潇予,丁哲旭,薛超,沈其荣
摘 要
网络分析逐渐被生态学家们重视并持续应用于生态学领域,开发更为强大和方便的网络分析工具十分必要。因此,我们开发了名为ggClusterNet的R包,用于更加容易的进行网络数据分析挖掘和可视化。在ggClusterNet包中设计了数十种网络布局算法用于更好的展示微生物网络模块化信息(randomClusterG, PolygonClus-terG, PolygonRrClusterG, ArtifCluster, randSNEClusterG, PolygonModsquar-eG, PolyRdmNotdCirG, model_Gephi.2, model_igraph, and model_maptree)。为了方便研究者使用,设计了多种微生物网络数据挖掘功能,例如相关性计算(corMicor()),网络属性计算(net_properties()),节点属性计算(node_properties()),随机网络计算和比对(random_Net_compate())。为了更加快捷挖掘网络,进一步将这些功能封装整合成两个函数network.2() 和 corBionetwork(),分别用于微生物网络数据挖掘和跨域网络数据挖掘。目前,ggClusterNet在github(https://github.com/taowenmicro/ggClusterNet/),Gitee(https://gitee.com/wentaomicro/ggClusterNet)上开放使用。完整的说明和示例在wiki页面可以阅读。
关键词:微生物组, 网络分析, R包, 可视化
亮 点

● ggClusterNet用于挖掘微生物网络和跨域网络,同时提供数十种基于模块化展示的网络布局算法,适应微生物组网络挖掘
视频解读
Bilibili:https://www.bilibili.com/video/BV1ig411Q7cw/
Youtube:https://youtu.be/Kxi_pn4QJrU
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
微生物组网络分析方法发展和现状
网络分析不仅在探索一组项目(节点)以及它们之间连接(边)的数学、统计和结构属性中具有重要作用,而且也被广泛用于探索复杂群落内微生物分类群之间的共现模式。例如,马斌等人基于网络分析发现不同环境中微生物组的互作模式,并强调了网络分析对微生物组群落分析的重要性;Yuan等人通过网络分析发现气候变暖增强了微生物网络的复杂性和稳定性;Franciska等人采用网络分析发现干旱会对土壤细菌网络产生巨大扰动,而对真菌网络波动影响较小。
微生物组网络分析的工具主要包括网页分析工具MENA(MENAP)、R软件包(WGCNA、igraph、ggraph、SpiecEasi)、交互式生物软件(Cytoscape和Gephi)、python模型(NetworkX和SparCC)等。有关网络相关计算的工具主要有:MENA、WGCNA、SpiecEasi等。MENA可以完成微生物网络相关矩阵计算,基于随机矩阵理论(RMT)方法,对噪声具有鲁棒性;WGCNA用于构建基于软阈值的无标度拓扑加权网络,通过到节点之外的其他相关关系加权更加具有生物学意义;SpiecEasi为数据开发的数据转换与图形模型推理框架相结合,生成了一套计算工具,可以有效解决少样本的鲁棒性问题,降低相关性计算过程中的假阳性。网络可视化的工具主要有Cytoscape、Gephi和R包(igraph、ggraph等)。Cytoscape不仅可以提供强大的可视化方案,而且允许用户通过多种插件开发新的功能,可以完成微生物网络挖掘等;Gephi可以通过少量的操作步骤轻松完成美观的网络图形。R软件包中的可视化工具(igraph、ggraph)也可以在命令行中快速展示网络,并且具有较高可重复性。但是,这些工具虽然在网络分析的不同方面具有优势,但是仍然不能满足我们对于快速计算,可重复性,深入挖掘的网络需求。例如,Cytoscape的步骤繁琐,不容易重复,而igraph和ggraph的布局不够美观。
目前,更多的研究人员关注微生物网络模块之间的相互作用,来探索它们的功能。然而,网络中各种微生物物种之间的联系太多,而且缺乏合适的可视化方案和软件足够清晰的展示模块之间的关系,使得这些工具在实际使用中不足以满足当下相关科研工作者的需求。为了解决网络分析中日益增长的需求,我们通过R语言开发了R包ggClusterNet。它为网络分析提供了一个快速、可重复和易于使用的流程,并具有多种强大的基于模块化的可视化布局。ggClusterNet具有以下特点:1)网络分析流程可以快速完成;2)网络分析流程可以提供多种精美的可视化布局;3)网络分析流程只需少量代码即可完成,极具重复性。
ggClusterNet开发环境和过程介绍
ggClesterNet是基于R语言开发的网络分析流程工具。其中,网络计算使用的工具主要调用了cor()函数(R包WGCNA),sparccboot()函数(R包SpiecEasi)和corr.test()函数(R包psych)。部分网络布局参考了ggraph和sna包。网络属性计算工具主要调用了igraph包中函数(average.path.length()、edge_connectivity()、no.clusters()、centralization.closeness()、erdos.renyi.game()等)。ggClusterNet软件开源,并可以在Github下载使用(https://github.com/taowenmicro/ggClusterNet/),也可以通过R语言命令行工具进行安装(devtools::install_github("taowenmicro/ggClusterNet"))。
为了丰富ggClusterNet对于模块化布局的支持,这里开发了超过10种基于模块化的网络可视化布局函数,这些函数分别是:randomClusterG、PolygonClusterG、PolygonRrClusterG、ArtifCluster、randSNEClusterG、PolygonModsquareG、PolyRdmNotdCirG、model_Gephi.2、model_igraph和model_maptree。这些算法需要相关矩阵和节点的模块化信息作为输入,可以计算各种样式的网络布局。这些布局结果最后可以使用R语言ggplot2包进行可视化。
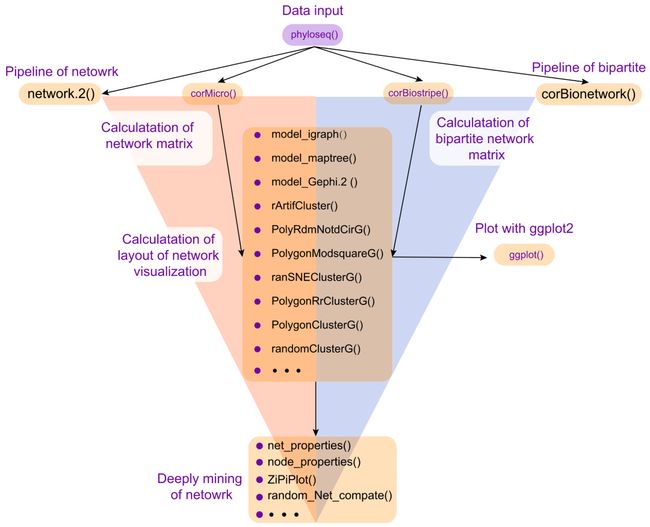
图1. ggClusterNet功能和函数介绍
ggClusterNet的使用
● ggClusterNet的工作流程
在ggClusterNet中,corMciro()(用于微生物网络的相关矩阵计算)或corBiostripe()(用于二分网络的相关矩阵计算)被用来计算相关矩阵。设计了十多种网络布局算法来计算相关矩阵的可视化的布局,计算结果并用ggplot2进行绘制。分别使用net_properties(),node_properties()和ZiPiPlot()来计算网络属性、节点属性,以及根据模块计算节点的作用。使用random_Net_compate()计算零模型并生成随机网络,并比较和微生物网络中的网络属性。以上这些函数都包含在nerwork.2()(微生物组网络流程)或corBionetwork()(二分网络的流程),可以通过一条函数运行网络分析全部内容。总之,ggClusterNet可以快速完成整个微生物组和二分网络的分析,包括相关性计算、网络可视化、网络属性计算、节点属性以及随机网络的构建和比较。

图2. ggClusterNet函数使用和输出结果介绍
● 网络布局
为了更好地实现微生物网络的可视化并突出模块互作关系,开发了十种可视化的布局算法。下面介绍这些算法的功能。
1)randomClusterG:将单个模块(组)的节点全部排列成一个环,然后将多个模块绘制成多个相同半径的圆圈并在绘图面板上随机地排列这些圆环。
2)PolygonClusterG:将单个模块(组)的节点排列成一个圆环。这些多个模块被绘制成多个相同半径的圆。最后这些代表不同模块的圆环被排列到同等数量边的多边形顶点上。
3)PolygonRrClusterG:单个模块(组)的节点都被排列成一个环。按照模块中节点数量将模块绘制成多个不同半径的圆(节点数越多,半径的越大)。然后,这些圆被有规律地排列到以坐标轴为中心的多边形的顶点上(边的数量等于模块的数量)。
4)ArtifCluster:单个模块(组)的节点都被排列成一个半径相同的圆环。通过设置坐标值,手动排列这些圆圈。
5)randSNEClusterG:单个模块(组)的节点可以调用sna包中的可视化布局计算坐标,不同模块随机排布到绘图面板上。
6)PolygonModsquareG:单个个模块(组)的节点都被安排成一个圆环。不同模块被绘制成多个不同半径的圆圈(节点数越多,半径越大)。这些圆圈通过少量参数排列成一行或多行。
7)PolyRdmNotdCirG:根据模块信息,将节点随机分布在多个不同半径的圆内(节点数越多,半径越大)。将这些模块有规律地排列到以坐标轴原点为中心的多边形的顶点上(边的数量等于模块的数量)。
8)model_Gephi.2:所有的节点计算出坐标并排布成一个圆。使用微生物聚类结果重新分配坐标。
9)model_igraph:使用Fruchterman和Reingold的算法,将所有节点按照模块分类投射到坐标轴上。
10)model_maptree:首先对网络进行了模块化分析,并按网络模块化程度对节点进行分组,然后用于计算坐标。节点的相对位置是根据王维新等人开发的算法计算,该算法试图按照模块化信息对同一模块节点趋向于映射到坐标轴的相近位置。
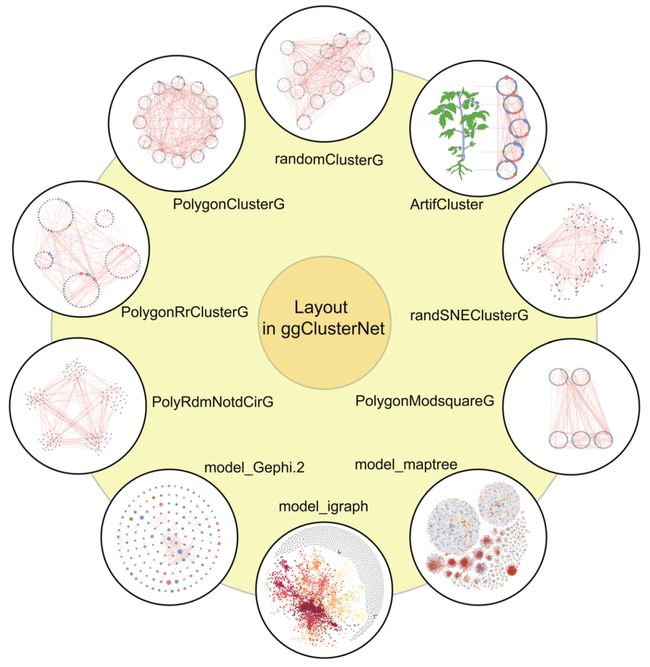
图3. ggClusterNet中的可视化布局
ggClusterNet功能特性和未来开发方向
网络分析日益受到微生物生态学家的青睐。近些年来,开发了许多功能强大的工具,如Cytoscape、Gephi、igraph等。在可视化方面,Cytoscape和Gephi因其交互式的图形用户界面和有吸引力的可视化结果而闻名。Cytoscape为网络分析提供了强大的功能,但需要调整许多参数来实现网络中的模块可视化。Gephi可以使用默认参数快速展示网络中的模块,而对网络的深度挖掘不足。ggClusterNet综合了igraph、ggraph、sna的优点,提供了多种布局算法,并且提供流程函数(network.2()和network())可以快速展示网络中的模块并对网络进行深度挖掘。
未来的工作将继续开发优化ggClusterNet包。为了增强微生物组网络分析的功能,将在流程中加入网络的稳定性内容,并深度挖掘模块的生态学功能;使用Shiny构建用户友好的界面,方便更多的研究者对网络进行深入探索;此将进一步挖掘二分网络的分析,开发更多适合二分网络的可视化布局算法。总之,ggCLusterNet希望不断完善网络分析相关内容,并帮助广大相关的科研工作者。
引 文
Tao Wen, Penghao Xie, Shengdie Yang, Guoqing Niu, Xiaoyu Liu, Zhexu Ding, Chao Xue, Yong-Xin Liu, Qirong Shen, Jun Yuan. 2022. ggClusterNet: An R package for microbiome network analysis and modularity-based multiple network layouts. iMeta 1: e32. https://doi.org/10.1002/imt2.32
作者简介

文涛(第一作者)
● 南京农业大学中山青年研究员,iMeta期刊青年编委
●专注土传病害微生物过程研究,擅长使用各种生物信息工具解决生态等问题。开发了ggClusterNet, EasyStat等R包, EasyAmplicon, EasyMetabolome等组学分析流程。在iMeta、ISME、Micribiome、Fundamental Research、Horticulture Research、SEL、BMC Plant Biology等期刊上发表了多篇文章。

袁军(通讯作者)
● 南京农业大学副教授,院士助理。研究方向集中于根际代谢物介导根际互作过程。
● 主要研究内容包括:1. 土传病害发生过程的植物和微生物互作;2. 环境微生物大数据整合研究;3. 根际微生态调控机理及技术开发 。以第一作者或通讯作者在 ISME J,Microbiome,Fundamental Research,iMeta, PCE,SBB,Horticulture Research,SEL,BMC plant biology等期刊上发表了二十余篇文章,被引超过1500次。
更多推荐
(▼ 点击跳转)
iMeta文章中文翻译+视频解读
iMeta封面 | 宏蛋白质组学分析一站式工具集iMetaLab Suite(加拿大渥太华大学Figeys组)

▸▸▸▸
iMeta | 华南师大王璋组综述人体肺部微生物组与人类健康和疾病之间的隐秘关联
▸▸▸▸
iMeta | 南科大夏雨组纳米孔测序揭示微生物可减轻高海拔冻土温室气体排放
▸▸▸▸
iMeta | 北大陈峰/陈智滨等发表口腔微生物组研究中各部位取样的实验方法(Protocol)

▸▸▸▸
iMeta | 华南农大曾振灵/熊文广等-家庭中宠物犬与主人耐药基因的共存研究
▸▸▸▸
iMeta | 深圳先进院马迎飞组开发基于神经网络分析肠道菌群的方法
▸▸▸▸
iMeta | 南医大陈连民等综述从基因组功能角度揭示肠菌对复杂疾病的潜在影响

▸▸▸▸
iMeta | 北大陈峰组综述口腔微生物组的标准化研究:从技术驱动到假说驱动
▸▸▸▸
iMeta | 电子科大林昊组开发蛋白质赖氨酸乳酸化位点预测工具DeepKla

▸▸▸▸
iMeta | 中科院李小方等膳食甘草促进小鼠镉解毒并调节肠道菌群代谢

▸▸▸▸
iMeta | 浙大倪艳组MetOrigin实现代谢物溯源和肠道微生物组与代谢组整合分析
期刊简介

“iMeta” 是由威立、肠菌分会和本领域数百位华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表原创研究、方法和综述以促进宏基因组学、微生物组和生物信息学发展。目标是发表前10%(IF > 15)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!
联系我们
iMeta主页:http://www.imeta.science
出版社:https://onlinelibrary.wiley.com/journal/2770596x
投稿:https://mc.manuscriptcentral.com/imeta
邮箱:[email protected]
微信公众号
iMeta
责任编辑
微微
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























机器学习
后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


