A Deep Generative Model for Molecule Optimization via One FragmentModification 1 【整体理解】
Modof:A Deep Generative Model for Molecule Optimization via One FragmentModification
基于单片段修改的分子优化深度生成模型
paper and supplement metrics : A deep generative model for molecule optimization via one fragment modification | Nature Machine Intelligence
data and code: GitHub - ninglab/Modof: The implementation of Modof for Molecule Optimization
文章介绍了一种新的深度生成模型Modof,它预测给定分子的单个断开位点,通过在该位点去除和/或添加片段进行分子修改。Modof每次只修改一个片段,保留了主要分子支架,可控制中间优化步骤并能更好的约束分子相似性。
以往的研究工作通常是对整个分子图进行编码,然后从一个空的或随机选择的结构中生成新的分子。不同的是,Modof是学习成对分子之间的差异并对差异进行编码和解码,学习和生成过程较为简洁,且能够保留主要的分子支架。
模型方法
训练集:作者采用Jin等人从ZINC数据集中提取的75K对基准训练集(其中每对分子结构相似,但plogP值不同)。使用DF-GED算法,作者又从该训练数据集中提取了55686对分子,其中每对分子都有一个断开位点nd,使用这些分子对(包含104708个独特的分子)作为初始训练数据。
测试集:使用800个成对的分子。
对比的SOTA模型有:
- JT-VAE对结树进行编码和解码,并基于解码的结树组装新的整个分子图。
- GCPN应用图卷积策略网络,通过逐个添加原子和键来迭代生成分子。
- JTNN从分子对中学习,并进行分子优化,以翻译分子图。
- HierG2G以分层的方式编码分子图,并通过生成和连接结构基序来生成新的分子。
- GraphAF学习先验分布和分子结构之间的可逆映射,并使用强化学习来微调模型以进行分子优化。
- MoFlow学习键邻接张量与高斯分布之间的可逆映射,然后应用图条件流从映射中生成一个原子型矩阵作为一个新分子的表示。
Modof 具有以下优点:
- 它一次修改一个片段。它更好地接近体外化学修饰并保留了大部分分子支架。因此,它可能更好地告知和指导体外分子优化。
- 它只对需要修改的片段进行编码和解码,有利于更好的修改性能。
- Modof-pipe 迭代地修改不同断开站点的多个片段。它可以更轻松地控制和直观地破译中间修改步骤,并有助于更好地解释整个修改过程。
- Modof 没有最先进的技术那么复杂。它的参数至少减少了 40%,使用的训练数据减少了 26%。
- Modof-pipe 在优化辛醇-水分配系数方面优于基准数据集上的最先进方法,该系数受到合成可及性 (SA) 和环尺寸的影响,在优化分子没有分子相似性约束的情况下提高了 81.2%,而优化了 51.2 %、25.6% 和 9.2% 的改进,如果优化后的分子需要至少与优化前的分子相似,分别为 0.2、0.4 和 0.6。
- Modof-pipem 将 Modof-pipe 的性能提高了至少 17.8%。
- Modof-pipem 和 Modof-pipe 在其他两项基准测试任务中也表现出卓越的性能,优化分子对多巴胺 D2 受体的结合亲和力并改善通过定量测量估计的药物相似性
3.问题定义
定义:给定分子Mx,将Mx修改成另一个分子My,My满足下面的条件:
(1)相似性约束:My的分子结构与Mx相似,即sim(Mx, My)≥δ(δ是一个阈值);
(2)属性约束:My的分子结构特征优于Mx,例如plogP(My)>plogP(Mx),其中plogP表示logP、可合成性(SA)和环数量的组合测量值。
分子相似性计算
通过 Wildman 和 Crippen 方法估计 Crippen logP
使用预定义片段的评分函数计算合成可访问性SA,使用支持向量机分类器预测 DRD2 属性
利用非线性分类器结合分子性质的多个期望函数对量子电动力学性质进行了预测
作者使用半径为2的2048维摩根指纹来表示分子,并使用Tanimoto系数(谷本相似度)计算分子相似性。
5.方法
符号表示:

M = (G, T)
G = (A, B)
J = (V, E)
首先,这里的分子embedding有两种形式,分别是graph的MPNN框架下的GMPN( Gx , Gy )和junction tree的TMPN( Tx , Ty )。其中, Gx=(Ax,Bx) 是低活性的分子, Ax 是原子类型, Bx 是原子相应的边。 Tx=(Vx,Ex) , Vx 是分子中所有的环和键作为节点, Ex 是具有共同原子的节点相连的边。
输入:我们在 Modof 中使用了一对分子(Mx,My)作为训练实例,其中 Mx 和 My 同时满足相似性和属性约束,而 My 与 Mx 的不同之处仅在于一个断开位点的一个片段(每对分子相似,而且一个属性优于另一个,且仅具有一个断开位置)。
Modof一次只修改分子的一个片段,因此只对需要修改的片段进行编码和解码,框架如图3所示。
Modof-encoder:
- Modof使用图消息传递网络(GMPN)在分子图/上生成 Mx/My的原子嵌入
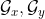 ,
, - 并使用树消息传递网络(TMPN)在相应的连接树/上生成节点嵌入
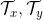
- 利用Difference Embedding (DE)学习得到“节点嵌入 ”断开点的差别,并将这部分差异的地方取出来,分别嵌入为
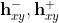 ,
, - 再将 映射为符合正态分布的隐向量
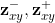 。
。
Modof-decoder:
- 将
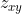 ()当做输入,然后Modof使用"断开位点预测(DSP)"来识别断开位点
()当做输入,然后Modof使用"断开位点预测(DSP)"来识别断开位点 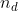 (片段被修改的位点称为断开位点并表示为 ,它对应于连接树表示中的一个节点)
(片段被修改的位点称为断开位点并表示为 ,它对应于连接树表示中的一个节点) - 在nd的邻域,Modof进行“移除片段预测 (RFP)”以移除nd连接的某个片段
- 然后,Modof使用“中间表示(IMR)”保留剩余scaffold(
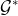 ,
,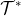 )。
)。 - 在 ( ,)上,Modof通过迭代三种“新片段附着(NFA)”执行"子节点连接预测(NFA-cp)"、"子节点类型预测(NFA-ntp)"和"附着点预测 (NFA-app)" 来实现Mx的优化。
6.实验结果
plogP优化整体比较
表1给出了Modof-pipe、Modof-pipem和其他基准方法的比较。当δ=0时,即相似性无约束,Modof-pipe能够产生优化效果更好的分子,plogP显著提高(7.61±2.30),比最佳基线GCPN(4.20±1.28)提高81.2%;Modof-pipem的plogP提高到了9.37±2.04,比最佳基线GCPN提高了123.1%。当约束分子生成前后的相似性时(即δ=0.2,0.4和0.6),基线比Modof-pipe产生更多相似的分子,但Modof-pipe的plogP性能优于基线方法,较最佳基线分别提高了51.2%、25.6%和9.2%;Modof-pipem的性能改善效果最佳,分别较最佳基线改善84.0%、48.0%和26.1%。
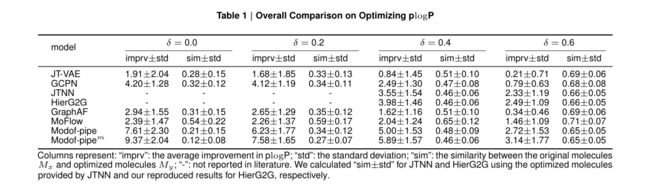
表1 plogP优化总体比较
Modof pipeline
它是由一连串相同的Modof models构成的,上一个Modof models的输出是下一个Modof models的输入:
- 给第 t 个Modof models一个分子输入M(t),Modof 首先优化 M(t) --> M(t + 1),其中 M(t + 1)是该第t个Modof models的输出
- 如果满足相似性 sim(M(t+1),M)>δ 以及性质相似性 plogP(M(t+1))>plogP(M(t) ), M(t + 1) 就会被输入到第 t+1 个Modof models,否则 M(t) 就是最后的结果,然后 Modof-pipe 结束
- 更容易控制中间优化步骤,从而得到具有所需相似性和性质的优化分子;
- 更容易优化一个分子中相距很远的多个片段
- 它遵循一个合理的分子设计过程,因此可以获得更多的见解,并为体外先导优化提供信息。
Model Training
在模型训练过程中,我们采用教师强迫的方法将预测结果输入到序列解码过程中,参考AE,我们最小化如下损失,并且最大化可能性 ![]() ,因此最终的优化问题被定义为:
,因此最终的优化问题被定义为:
![]()
- 其中
是参数设置,
() 是估计的后验概率(Modof - encoder),
是概率 decoder,表示在给定潜在嵌入 z 和
的情况下生成
的可能性,其中先验
服从 正泰分布 N(0,1)
() 是 p() 和 q() 之间的KL散度,
- 第二项为预测或经验误差,定义为以上的损失函数和,作者使用AMSGRAD来优化目标函数
案例研究
训练的分子中,从Mx上删除的前5个片段及其规范的SMILE字符串显示在图1a中;要附加在生成的My(优化后的分子)的前5个片段显示在图2b中。总的来说,训练数据中的去除片段平均为2.85个原子,新片段平均为7.55个原子。也就是说,优化通常是去除小片段然后附加较大的片段。
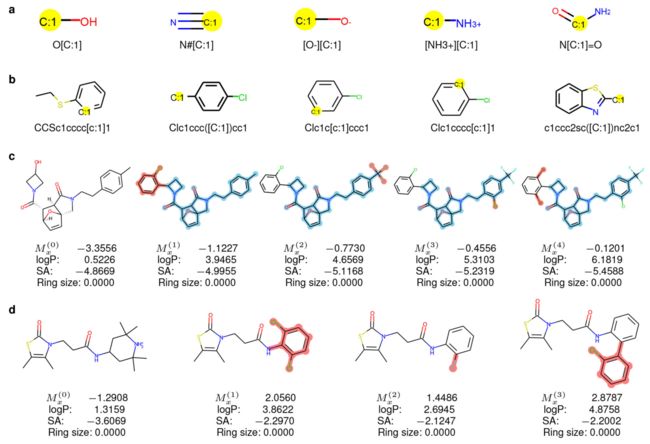 图1 Modof-pipe优化plogP示例
图1 Modof-pipe优化plogP示例
- a:可视化常用的删除片段
- b:可视化常用的添加片段
- c:Modof-pipe 优化示例:多个断开点和多个Modof迭代
- d: 局部优化
其中:disconnection的点高亮为黄色,每一次改变的片段为红色,在Modof-pipe优化后还保留的高亮为天空蓝色,Mx’s对应的值是plogP值
图1c展示了一个分子Mx(即Mx(0)),δ=0.4时在Modof-pipe中通过四次迭代优化成另一个分子Mx(4)。在每次迭代中,只有一个小片段被修改(图中红色部分),plogP值得到提高。在第一次迭代中,Mx(1)是通过去除Mx(0)中的羟基和添加2-氯苯基得到。羟基是极性的,往往会增加分子的水溶性,而2-氯苯基是非极性的,因此会增加分子的疏水性。此外,氯苯取代基带来的分子量增加也降低了水溶性。因此,从羟基到氯苯基的修改使logP从0.5226增加到3.9465。同时,在环丁基上引入2-氯苯基增加了合成的复杂性,加上芳香环上邻位取代可能造成的空间效应,导致SA从−4.8669下降到−4.9955。在第二次迭代中,Mx(1)中的甲基被三氟甲基取代。三氟甲基比甲基更疏水,因此使得Mx(2)的logP值超过Mx(1)(从3.9465到4.6569)。同时,较大的分子Mx(2)的SA值也比Mx(1)更差(从-4.9955到-5.1168)。如果按照Lipinski的“五倍律”,当logP值小于5,则可以停止Modof-pipe的迭代;否则,在接下来的两次迭代中,更多的卤素被添加到芳香环上,这会使芳香环极性降低,进一步降低水溶性并增加logP值。这些迭代突出了Modof-pipe对应于化学知识的可解释性。值得注意的是,Modof中的所有修改都是从数据中以端到端的方式学习的,没有任何先验的化学规则或模板,强调了Modof学习能力的强大。
DRD2和QED的性能优化
除了改善plogP之外,分子优化的另外两个常用理想特性是提高“分子对多巴胺D2受体的结合亲和力(DRD2)”,以及提高“定量评估的药物相似性(QED)”:具体来说,给定一个不能很好地结合DRD2受体的分子,优化DRD2特性的目标是将分子修改成另一个能更好地结合DRD2的分子。在QED任务中,给定一个不太像药物的分子,优化QED特性的目标是将这个分子修改成一个更像药物的分子。
表2给出了相似约束δ=0.4下的成功率、性能改进和相似度比较。结果表明,当使用基准指标(表2的OM-pic)或基于训练数据(表2的OM-trn)测量成功率时,Modof-pipem在优化DRD2和QED方面效果更好,可与基线方法相媲美。图3a和图3b给出了两个分子优化DRD2和QED的例子。作者还进行了同时优化分子DRD2和QED性质的实验,即多目标优化任务。图3c是一个分子多目标优化的例子,在这个例子中,分子的DRD2和QED分数都随着优化的迭代不断增加。
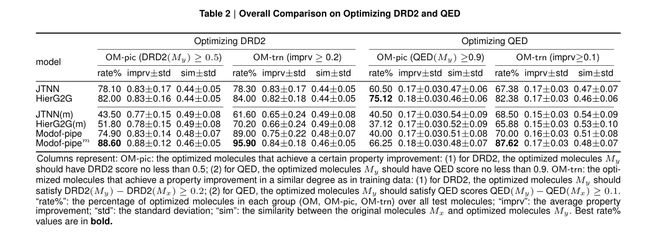
表2 DRD2与QED优化的总体比较
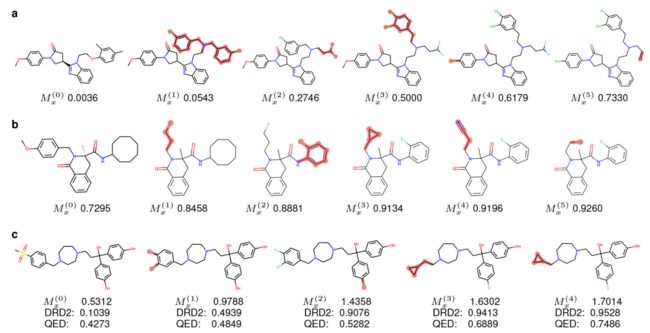 图3 Modof-pipe DRD2, QED 和 multi-property 优化示例
图3 Modof-pipe DRD2, QED 和 multi-property 优化示例
- a:DRD2 的Modof-pipe 优化示例
- b: QED 的Modof-pipe 优化示例
- c: DRD2 and QED 的multi-property优化示例
改变的部分高亮为红色
7.讨论和结论
Modof局限性
(1)作者在实验中考虑的分子性质是基于模拟值或预测值,而不是通过实验测量获得【也就是只是干试验,没有湿实验】。虽然现有的分子优化生成模型都使用这些模拟属性,但当这些预测由于各种原因不够准确时(例如,受限或有偏差的训练分子)就会出现问题,导致生成的分子可能会对药物开发任务产生显著的负面影响。
(2)实验表明,Modof能够提高分子的SA性能,但这并不意味生成的分子可以很容易地合成。Modof的这种局限性存在于大多数分子生成模型中。这种合成能力的缺乏,极大地限制了生成模型在药物开发中产生实际影响的潜力。
(3)采用了局部贪心优化策略,在每次迭代中,输入到Modof的分子会被优化到最优,如果优化后的分子没有更好的性质,将不会进行下一次Modof迭代。
未来展望
(1)深度学习的逆合成预测是一个活跃的研究领域,它的目的是通过学习和搜索大量的合成路径,为给定的分子找到一条可行的合成路径。优化分子,使其不仅具有更好的性能,而且具有更好的合成能力,特别是还要同时确定明确的合成路径,可能是未来一个非常有趣且具有挑战性的研究方向。
(2)与制药行业和体外试验合作,在实验室中测试计算机辅助生成的分子,将计算方法转化为真正的成果。计算机生成的分子的大规模体外验证代表了一个未来研究的方向。
(3)增强Modof修改分子的内部区域的能力。此外,作者还希望在Modof学习过程中整合特定领域的知识,以提高模型在学习和生成过程中的可解释性。
Nat. Mach. Intell. | 基于单片段修改的分子优化深度生成模型 - 知乎
论文:A Deep Generative Model for Molecule Optimization via One FragmentModification_m0_47163076的博客-CSDN博客_graphvae论文中文
使用深度学习,通过一个片段修饰进行分子优化 - 知乎
A Deep Generative Model for Molecule Optimization via One Fragment Modification论文笔记 - 知乎