【学习笔记AProt】
一. 代码
https://github.com/arontier/A_Prot_Paper
二. 摘要
背景:借助深度学习的进步,蛋白质3D结构预测的准确性得到了显著提高。在最近的CASP14中,Deepmind证明了他们新版本的AlphaFold(AF)可以产生几乎接近实验结构的高精度3D模型。AF的成功表明,序列的多序列对齐包含丰富的进化信息,从而导致准确的3D模型。尽管AF取得了成功,但只有预测代码是开放的,训练类似的模型需要大量的计算资源。因此,开发一个更轻的预测模型仍然是必要的。结果:在本研究中,我们提出了一种新的蛋白质3D结构建模方法,即A-Prot,使用最先进的蛋白质语言模型之一MSA变压器。提取MSA特征张量和行注意图,并将其转换为给定MSA的二维残留距离和二面角预测。我们证明,A-Prot比现有方法更好地预测远程接触。此外,我们还对CASP14的自由建模和基于硬模板的建模目标的3D结构进行了建模。评估表明,A-Prot模型比CASP14的大多数顶级服务器组更准确。结论:这些结果表明,A-Prot以相对较低的计算成本准确捕获蛋白质的进化和结构信息。因此,A-Prot可以为开发其他蛋白质特性预测方法提供线索。
结果:在本研究中,我们提出了一种新的蛋白质3D结构建模方法,即A-Prot,使用最先进的蛋白质语言模型之一MSA变压器。提取MSA特征张量和行注意图,并将其转换为给定MSA的二维残留距离和二面角预测。我们证明,A-Prot比现有方法更好地预测远程接触。此外,我们还对CASP14的自由建模和基于硬模板的建模目标的3D结构进行了建模。评估表明,A-Prot模型比CASP14的大多数顶级服务器组更准确
结论:这些结果表明,A-Prot以相对较低的计算成本准确捕获蛋白质的进化和结构信息。因此,A-Prot可以为开发其他蛋白质特性预测方法提供线索
关键词:蛋白质结构预测、多序列对齐、蛋白质语言模型、深度学习
三. 引言
从蛋白质序列中建模蛋白质的3D结构一直是生物物理学和生物化学中最关键的问题之一[1,2]。对蛋白质3D结构的了解有助于发现新的配体、功能注释和蛋白质工程。由于其重要性,计算蛋白质科学家社区一直在开发各种预测方法,并评估其在大规模盲目测试CASP中的表现,该测试已经持续了二十多年[2]。在CASP14中,Deepmind证明,他们基于深度学习的模型AlphaFold2(AF2)以极高的精度从其序列中预测蛋白质的3D结构,可与实验精度相当[3]。AF2的源代码最近公开发布,许多具有生物重要性的生物基因模型结构已经发布[3]。
AF2的成功可以归功于从多序列对齐(MSA)准确提取共进化信息。使用共进化信息进行接触预测或结构建模的想法已被广泛使用[4-12]。然而,之前的尝试不够成功,因为MSA中的共同进化信号不够强烈或太吵。提出了各种基于统计力学的模型,但其区分实际接触和虚假接触的准确性有限。在CASP13中,AlphaFold和RaptorX使用来自MSA的共进化信息作为输入,并预测了残留距离[13-15]。他们比以前的CASP取得了重大改进,但他们的预测不够准确,无法获得与实验相媲美的模型结构。最后,在CASP14中,借助注意力算法,可以从MSA中真正准确提取实际残留物-残留接触信号[3]
尽管AF2取得了这一显著成就,但该技术仍然可以广泛访问并用于进一步开发,但仍存在某些限制。首先,训练AF2的源代码尚未开源。在AF2的第一个版本中,只有生产、模型生成、AF2的一部分和模型参数是开放的。其次,训练AF2架构在计算上很昂贵。据报道,Deepmind使用128个TPUv3内核大约一周零四天来训练AF2。对于大多数学术团体来说,这些计算资源并不容易获得。因此,开发更轻、更通用的模型仍然是必要的
为了从MSA中提取进化信息,提出了各种蛋白质语言模型[16-22]。Rao等人提出了MSA变压器模型,这是一个使用查询序列的MSA而不是单个查询序列的非监督蛋白质语言模型。该模型使用输入MSA和屏蔽语言建模目标的行和列关注。事实证明,该模型成功预测了残留物之间的远距离接触。此外,该模型以高精度预测了蛋白质的其他特性,如二级结构预测和突变效应[20]。这些结果表明,MSA变压器模型有效地从MSA配置文件中提取蛋白质的特性
本研究开发了一种新的蛋白质3D结构预测方法,A-Prot,使用MSA变压器[22]。对于给定的MSA,我们使用MSA变压器提取了进化信息。将提取的行注意图和输入特征转换为二维残留距离图和二面角预测。我们使用CASP13和14[23、24]的FM/TBM硬靶点对3D蛋白质结构建模性能进行了基准测试。结果表明,在远距离接触预测和3D蛋白质结构建模方面,A-Prot的表现优于CASP13和14的大多数顶级服务器组。
四. 模型框架
1. 总览
拟议蛋白质3D结构预测方法的整体管道如图所示。1.我们主要结合了MSA变压器[22]和trRosetta[25]的作品。给定MSA,它将输入MSA变压器,以输出MSA功能和行注意图。在维度缩小和串联等一系列转换后,这两种特征将被转换并组合成二维特征图,适合输入trRosetta最终输出蛋白质3D结构
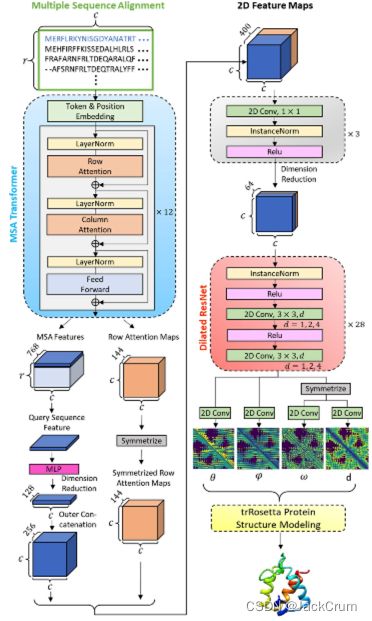
图1 拟议蛋白质3D结构预测方法的管道。将MSA输入MSA到MSA变压器,以提取MSA功能和行注意图。然后,通过一组转换将与查询序列和行注意映射相对应的MSA功能组合到2D特征映射中。接下来,在维度缩小后,将2D特征映射输入到扩张的ResNet,以输出残留之间的几何,这些几何进一步输入trRosetta蛋白质结构建模,以输出预测的蛋白质3D结构
本文中使用的MSA变压器是一个已经经过预训练的版本,从2600万个MSA中学习。它作为特征提取器发挥作用,输入MSA并输出其相对特征。
trRosetta由深层神经网络部分和蛋白质结构建模部分组成。对深度神经网络进行了修改,以接收MSA变压器的MSA功能,而不是统计方法生成的原始手动工程特征。然而,蛋白质结构建模部分保持不变。
2. 数据集和MSA生成
我们使用MSA变压器的程序生成目标序列的MSA(Rao,Liu等人,2021年)。查询序列的MSA是使用Jackhammer [26]生成的,HHblits ver.3.3.0和HHsearch [27]以及unicluster30_2017_10 [28]和BFD [29]数据库。如果没有指定,所有培训和测试MSA都将使用BFD。如果检测到的同源序列数量超过256个,则通过最小化选择多达256个序列[22]。序列数量的上限由NVIDIA Quadro RTX 8000 GPU(48 GB)卡的GPU内存的最大大小决定。
为了执行接触预测,编译了一个定制的非冗余蛋白质结构数据库,并将其命名为PDB30。PDB30数据集由2018年4月30日之前沉积在PDB中的蛋白质结构组成,其分辨率高于2.5 A,序列长度长于40个氨基酸。使用102,300个满足该条件的序列,使用MMSeq2 [30]进行了聚类分析,序列恒等式阈值为30%,导致16,612个非冗余序列。
3. 网络架构
让我们将输入MSA定义为r×c字符矩阵,其中r和c分别是MSA中的行(序列数)和列(序列长度)。通过MSA变压器的令牌和位置嵌入,输入MSA嵌入到r × c × 768张量中,这是每个注意力块的输入和输出。注意块按行注意层、列注意力层和前馈层的顺序组成。每个图层都遵循图层归一化操作。每个注意力层都有12个注意力头。MSA变压器是一堆12个这样的注意力块。
从MSA变压器中提取了两种特征,通过一系列变换来构建二维特征图。(1)一个是最后一个注意力块的输出,一个r × c × 768张量;我们将其命名为MSA功能(图1)。只选择与查询序列相对应的特征,即1×c×768张量。然后,MLP(多层感知器)将该特征的维度减少到128,该MLP(多层感知器)由3个线性层组成,神经元大小为384、192、128。然后,维度简化的1D特征被外部串联(水平和垂直冗余扩展,然后堆叠在一起),形成c × c × 256张量的查询序列特征。(2)另一个是从每行注意力层的每个注意力头派生出来的行注意力图,总共12×12 = 144张堆叠成c × c × 144形状的注意力地图。然后通过将其添加到其移调张量中来对称,以生成对称的行注意力映射。查询序列特征和对称行注意映射在特征映射维度中串联,形成c×c×400张量的二维特征图
2D特征映射的维度随后由三个卷积层减少到64,这些卷积层由256、128、64个大小为1×1[31]的内核组成,其中每个卷积层之后都有一个实例归一化和ReLU激活。然后将这个c × c × 64张量输入到扩张的ResNet,由28个剩余块[32]组成,每个块都有一个实例归一化,两个ReLU激活和两个卷积层,每个层有64个大小为3×3的内核。膨胀应用于通过残余块的卷积层循环,速率为1、2、4。在最后一个残余块之后,有四个独立的卷积层,具有25、13、25、37个大小为1×1的内核,每个核分别用于预测θ、φ、ω、d的残余间几何。有关我们使用相同设置的残差间几何形状的更多详细信息,请参阅参考文献[25]。最后,trRosetta蛋白质结构建模模块将根据残留几何信息预测和建模蛋白质3D结构
4. 培训和推理
在训练阶段,我们修复了MSA变压器中的参数。相比之下,使用RAdam优化器[33],对其他深度神经网络中的参数进行了批次大小为16的训练,梯度积累步骤,学习率为1e−3,计算了四个残余间几何目标[25]的分类交叉熵损失。残差间几何的基真值都离散成与相应卷积内核具有相同数量的垃圾箱(25个bins为θ,13个bins为φ,25个bins为ω,37个bins为d);每个bin被视为分类标签。总型号在NVIDIA Quadro RTX 8000 GPU(48 GB)上端到端训练了大约40个时代,大约需要五天
在训练期间应用MSA子采样策略,不仅用于将数据增强以训练鲁棒模型,也用于防止GPU在填充大型MSA时内存耗尽。我们随机选择MSA行,最多214/c,最低16行,尽管总是包括第一行中的查询序列。在训练过程中丢弃了超过1023种残留物的大型蛋白质。我们在推理阶段用256个序列对MSA进行了子采样,方法是添加平均哈姆距离最低的序列。我们用相同的输入进行了五次trRosetta蛋白质结构建模,并选择了能量最低的结构进行蛋白质结构相似性测量。
五. 结果和讨论
1. 基准接触预测
首先,我们使用CASP13 [24]的FM和FM/TBM目标对A-Prot的远程接触预测性能进行了基准测试。基准结果表明,我们模型的性能优于现有方法(表1)。我们比较了模型顶部L/5、L/2、远程接触预测(远程:残留物对≥24的序列分离)和其他现有方法的精度。其他方法的性能指标来自参考文献[34]。我们型号的顶级L/5、Top L/2和L接触精度为0.812、0.710和0.562,高于其他方法。例如,最先进的方法之一DeepDist以0.793、0.661和0.517的精度预测了顶部L/5、L/2和L。此外,与CASP13的AlphaFold预测相比,A-Prot在所有三个指标中的预测都比7-9%
表1 对应43个域的CASP13 FM和FM/TBM目标的接触精度(结果改编自DeepDist [34])

我们还比较了A-Prot与MSA-Transformer的接触预测准确性,MSA-Transformer是A-Prot的基线。在CASP13 FM目标的监督接触预测方面,远程Top L和Top L/5的性能分别为0.546和0.775(Rao、Meier等人,2021年)。A-prot的Top L和Top L/5的接触精度为0.539和0.785,与MSA Transformer(附加文件1:表S2)的接触精度相当。我们还研究了MSA通过预测结构与没有BFD并获得的用DeepMSA获得的MSA[35]对接触预测的影响。评估表明,不使用BFD会恶化模型的质量。Top L和Top L/5联系人的准确性分别下降了2.5%和4.0%。使用DeepMSA时,Top L和Top L/5的准确率分别下降了3.9%和2.0%。这些结果表明,考虑一个巨大的元基因组数据库有助于提高预测质量,但并不显著。
为了研究残余块的数量如何影响预测质量,通过更改残余块的数量进行了消融测试(附加文件1:表S4)。我们对4、16、28和40个残余块进行了接触和结构预测。接触预测结果表明,28个块的A-Prot计算得出了最佳的接触预测。有趣的是,使用更多的块,40块,降低了接触预测的准确性。剩余块(4和16)的减少导致接触预测结果明显更差。就模型准确性而言,拥有40个块的A-Prot获得了最高的TMscore,尽管28个块的接触预测最准确。这些结果表明,考虑到预测准确性和计算成本,具有28个块的A-Prot接近最佳模型。
表2 25个CASP14 FM、FM/TBM和TBM硬域模型结构的平均TM评分和lDDT
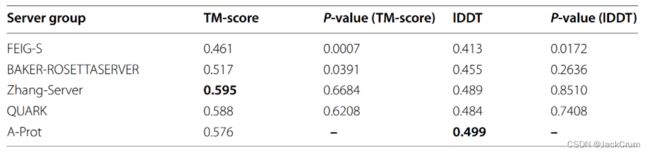
表3 A-Prot和trRosetta在25个CASP14 FM、FM/TBM、TBM-Hard域上使用与我们相同的MSA的性能。(使用最大概率预测的垃圾箱指数和地面真理之间的皮尔逊相关性测量残留间距离和角度,前L表示远距离接触精度)

2. 模型准确性的基准
除了接触预测外,我们还比较了A-Prot预测的蛋白质模型的质量与CASP14表现最好的服务器组提交的蛋白质模型的质量(表2)。每列的最高分数以粗体突出显示。首先,我们模拟了CASP14的25个FM/TBM和TBM硬目标的结构。将模型的平均TM评分和lDDT分数与以下服务器组进行比较:FEIG-S [36]、BAKER-ROSETTASERVER [37]、Zhang-Server和QUARK [38]。从CASP14网站的档案中下载了其他组的模型结构,并使用晶体结构和域信息重新计算了TM分数和lDDT分数,以便进行公平比较。
就lDDT而言,A-Prot的表现优于其他顶级服务器组。与BAKER- ROSETTASERVER和FEIG-S相比,A-Prot模型在这两种措施中都更加准确。P值表明,A-Prot在统计学上优于ROSETTASERVER和FEIG-S。这些结果表明,A-Prot的准确性与参与CASP14 [23]的性能最好的服务器组相当或更好。在TM得分方面,A-Prot显示的结果略差,为0.576,低于Zhang-Server和QUARK,后者的TM得分为0.595和0.588。然而,P值表明,A-Prot结果没有有意义的差异
我们还通过使用trRosetta与我们用于A-Prot的相同MSA建模结构,将A-Prot的性能与trRosetta[25]进行了比较(表3)。结果表明,使用相同的MSA,A-Prot的表现明显优于trRosetta。在二面角预测方面,A-Prot将地面真理和预测之间的相关系数平均提高了0.071。此外,A-Prot生成的模型比trRosetta结果具有更高的TMscore和lDDT值。平均TMscore和lDDT分别提高了0.048和0.042。
与MSA-Transformer或ROSETTA不同,A-Prot使用子采样策略,最大限度地减少序列的多样性,以减少MSA的大小。对于蛋白质语言模型,已经测试了四种子采样策略:随机、多样性最大化、多样性最小化和HF过滤器,以减少输入MSA的大小,同时保持预测准确性。结果表明,多样性最大化最适合监督接触预测[22]。相反,trRosetta以类似于多样性最小化方法[25]的方式生成MSA。在开发A-Prot期间,我们尝试了多样性最小化和最大化方法。预测结果表明,多样性最小化比多样性最大化(附加文件1:表S3)产生更准确的模型。我们使用具有多样性最小化和最大化的MSA子采样预测了43个CASP13 FM、FM/TBM域和25个CASP14 FM、FM/TBM和TBM硬目标域的结构。平均而言,使用多样性最小化获得的MSA生成的模型的TM得分更高,CASP13和CASP14目标的TM得分为0.658和0.555,高于多样性最大化的模型,即0.603和0.532。因此,我们采用了A-Prot的多样性最小化方法
3. 与ROSETTASERVER的正面比较
由于A-Prot在最后阶段使用trRosetta对结构进行建模,我们对A-Prot模型与ROSETTASERVER模型进行了正面比较,以确定残留距离预测的改进(图2)。就TM评分而言,A-Prot做出的许多预测明显优于BAKERROSETTASERVER。例如,ROSETTASERVER预测TM得分小于0.4的五个目标的模型质量高于0.4,对应于正确的折叠预测。图3 中描绘了四个高精度的模型。
同样,就LDDDT衡量标准而言,八个目标的预测结果得到了显著改善。相反,只有两个目标恶化了0.05以上。因此,A-Prot比大多数参与CASP14的服务器组更好地预测残留-残留距离和二面角[2]。


另一方面,除了T1026-D1目标外,A-Prot几乎所有更糟糕的预测都只恶化了很小的优势。对于T1026-D1,A-Prot没有预测其正确折叠。这种不正确的预测归因于MSA不完整。T1026-D1是一种由病毒衣壳(PDB ID:6S44)组成的蛋白质。我们的序列搜索过程只发现了不到30个序列,这似乎不足以提取正确的进化信息。当T1026-D1使用包含100多个序列的trRosetta的MSAT1026-D1建模时,可以获得具有类似TM分数的模型。因此,T1026-D1的故障表明,拥有足够的同源序列对于使用MSA变压器进行精确的3D结构建模至关重要。换句话说,更广泛的序列搜索可能会提高A-Pro的模型准确性。除T1026-D1外,所有比ROSETTASERVER更糟糕的预测偏差都小于0.1 TMscore,比改进小得多
六. 总结
在这项研究中,我们介绍了一种新的蛋白质结构预测方法,A-Prot,使用MSA变压器。我们对CASP13 TBM/FM和FM目标的基准结果表明,A-Prot比现有方法更准确地预测远距离残留接触。我们还根据预测的残留-残留距离信息评估了蛋白质结构模型的质量。A-Prot生成的模型比大多数参与CASP14的服务器组更准确。A-Prot模型的平均lDDT高于所有服务器组模型。就TM分数而言,我们的模型比QUARK和Zhang-Server略差。这些结果表明,我们的方法可以得出非常准确的残留-残留距离预测。
与其他最先进的蛋白质结构预测模型相比,A-Prot需要更少的计算资源[2]。AlphaFold2的源代码仅部分打开[3]。它的模型参数是固定的,只有结构建模部分是开放的。因此,很难为定制目的调整AlphaFold2。此外,培训AlphaFold2架构需要大量的计算资源,这对大多数学术团体来说是无法评估的。另一方面,A-Prot可以用一张GPU卡进行训练。总之,A-Prot将为训练基于深度学习的新型模型开辟新的可能性,仅使用序列信息预测蛋白质的各种特性。
缩写MSA:多序列对齐;FM:自由建模;TBM:基于模板的建模;RMSD:根均方偏差。
Abbreviations
MSA: Multiple sequence alignment; FM: Free modeling; TBM: Template-based modeling; RMSD: Root mean square
deviation.