R语言并行计算beta-NTI值
目录
- 一、beta-NTI(nearest taxon index)简介
- 二、系统发育信号(phylogenetic signal)检测
- 三、R代码并行实现beta-NTI计算
- 参考文献
一、beta-NTI(nearest taxon index)简介
1.首先简单描述一下下图一,这棵树就是遗传发育树,每个圈代表一个OTU,数字可认为是OTU ID,每个颜色代表一个样点,其中灰色是我们实验观察到的结果,红色和蓝色是我们进行1000次在发育树上随机分配的结果。
2.我们可根据实验观察和随机分配的结果计算MNTD(mean nearest taxon distance),然后将观察的结果与模拟的结果进行比较,得到NTI。
3.这里介绍的是样点内的NTI,可以理解为alpha-NTI,理解了alpha-NTI,再理解beta-NTI也是同样的道理(见图二),只不过是(2)中的MNTD换成了beta-MNTD。
4.一定要注意,只有OTU具有遗传发育信号(见图三),才能用beta-NTI去估算构建过程是决定性的还是随机的。
图一
 图二
图二 图三
图三
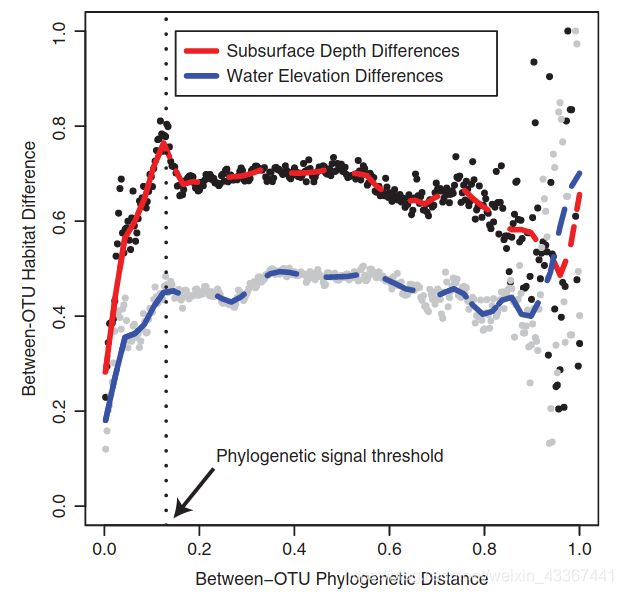
二、系统发育信号(phylogenetic signal)检测
步骤一:
大致判断OTU栖息地偏好性与OTU遗传发育距离之间的关系
phylosignal <- function(otu_niche,otu_tree,nclass){
library(picante)
library(ape)
prune_tree<-prune.sample(otu_niche,otu_tree)
tip<-prune_tree$tip.label
coln<-colnames(otu_niche)
m<-NULL
for(i in 1:length(coln)){
if(!coln[i]%in%tip){
print(coln[i])
m<-cbind(m,coln[i])
}
}
m<-as.vector(m)
otu_niche<-otu_niche[,!colnames(otu_niche)%in%m]
otu_phydist <- cophenetic(prune_tree)
#otu_bray<-vegdist(log1p(otu_niche), method="bray")
otu_bray<-vegdist(t(otu_niche), method="bray")
otu_bray<-as.matrix(otu_bray)
length(otu_bray) == length(otu_phydist)
#mntd(otu_niche,cophenetic(prune_tree))
otu_bray <- otu_bray[colnames(otu_phydist),colnames(otu_phydist)]
data1 <- data.frame(cor = c(unlist(otu_bray)),phydist=c(unlist(otu_phydist)))
colnames(data1) <- c("sorted_otu_bray_3","sorted_otu_phydist_3")
data1<-data1[order(data1$sorted_otu_phydist_3),]
fac1<-cut(data1$sorted_otu_phydist_3,nclass)
data2<-cbind.data.frame(fac1,data1)
library(plyr)
data3<-ddply(data2,.(fac1),colwise(median))
plot(data3$sorted_otu_phydist_3,data3$sorted_otu_bray_3)
physin <- data.frame(phydist=data3$sorted_otu_phydist_3,habpre=data3$sorted_otu_bray_3)
physin2 <- list(physin,otu_bray,otu_phydist)
names(physin2) <- c("physin","otu_bray","otu_phydist")
return(physin2)
}
步骤二:mantel correlogram
physin <- phylosignal(otu_niche,otu_tree,600)
physin$otu_bray[upper.tri(physin$otu_bray, diag=TRUE)] = NA
physin$otu_phydist[upper.tri(physin$otu_phydist, diag=TRUE)] = NA
man2 <- mantel.correlog(physin$otu_bray,physin$otu_phydist)
plot(1:6,man2$mantel.res[,5],lty = 1,type = "b")
abline(h = 0.05,lty = 5)
三、R代码并行实现beta-NTI计算
#本函数依赖picant、ape、doParrallel、foreach四个包
#threads:使用的线程数,可用detectCores()函数查看可使用的CPU数。
#使用的CPU数并不是越多越好,还要顾及自己的内存是否足够,因为每开一个线程都会占据相同的内存。
#2021年7月6日
beta_nti <- function(otu_niche,otu_tree,reps,threads){
library(picante)
library(ape)
prune_tree<-prune.sample(otu_niche,otu_tree)
tip<-prune_tree$tip.label
coln<-colnames(otu_niche)
m<-NULL
for(i in 1:length(coln)){
if(!coln[i]%in%tip){
#print(coln[i])
m<-cbind(m,coln[i])
}
}
m<-as.vector(m)
otu_niche<-otu_niche[,-which(colnames(otu_niche)%in%m)]
otu_phydist <- cophenetic(prune_tree)
match.phylo.otu = match.phylo.comm(prune_tree, otu_niche)
#str(match.phylo.otu)
## calculate empirical betaMNTD
#(算betamntd的)
beta.mntd.weighted = as.matrix(comdistnt(match.phylo.otu$comm,cophenetic(match.phylo.otu$phy),abundance.weighted=T));
#identical(rownames(match.phylo.otu$comm),colnames(beta.mntd.weighted)); # just a check, should be TRUE
#identical(rownames(match.phylo.otu$comm),rownames(beta.mntd.weighted)); # just a check, should be TRUE
# calculate randomized betaMNTD
beta.reps = reps; # number of randomizations
rand.weighted.bMNTD.comp = NULL
dim(rand.weighted.bMNTD.comp);
library(abind)
arraybind <- function(...){
abind(...,along = 3,force.array=TRUE)
}
library(foreach)
library(doParallel)
registerDoParallel(cores = threads)
rand.weighted.bMNTD.comp <- foreach (rep = 1:beta.reps, .combine = "arraybind") %dopar%{
#print(c(date(),rep))
as.matrix(comdistnt(match.phylo.otu$comm,taxaShuffle(cophenetic(match.phylo.otu$phy)),abundance.weighted=T,exclude.conspecifics = F))
}
weighted.bNTI = matrix(c(NA),nrow=nrow(match.phylo.otu$comm),ncol=nrow(match.phylo.otu$comm));
dim(weighted.bNTI);
for (columns in 1:(nrow(match.phylo.otu$comm)-1)) {
for (rows in (columns+1):nrow(match.phylo.otu$comm)) {
rand.vals = rand.weighted.bMNTD.comp[rows,columns,];
weighted.bNTI[rows,columns] = (beta.mntd.weighted[rows,columns] - mean(rand.vals)) / sd(rand.vals);
rm("rand.vals");
};
};
rownames(weighted.bNTI) = rownames(match.phylo.otu$comm);
colnames(weighted.bNTI) = rownames(match.phylo.otu$comm);
return(weighted.bNTI)
}
参考文献
[1] 原核微生物群落随机性和确定性装配过程的计算方法
[2] Stochastic and deterministic assembly processes in subsurface microbial communities
测试文件请参考付费资源,两者内容相同
面包多:https://mianbaoduo.com/o/bread/YpmTlZhx
CSDN:https://download.csdn.net/download/weixin_43367441/22468236
更多R语言分析微生物生态学的资源可参考如下链接:
https://mbd.pub/o/bread/mbd-YpmTlZpr