模型实践 | Alphafold 蛋白质结构预测
实验|Vachel 算力支持|幻方AIHPC
要说去年一年AI领域的最重大进展,DeepMind在Nature上发表的“AlphaFold”无出其右,其旨在根据一个蛋白质的氨基酸序列来确定它的3D结构,是生物学领域中的几个重大挑战课题之一。在“蛋白质结构预测关键评估”(CASP)的蛋白质结构预测年赛上,AlphaFold击败了其他百来支团队并取得了惊人的效果,有些时候,AlphaFold预测的结构与利用X射线晶体学和近几年的冷冻电镜(cryo-EM)等“金标准”实验方法所确定的结构几乎一样。科学家说,目前看来,AlphaFold暂时还不能取代这些费力又昂贵的实验方法,但它将带来全新的研究生命的方式。
作为一个计算机专业的算法从业人员,本期笔者带着浓厚的兴趣来尝试一下DeepMind开源的Alphafold模型并验证效果。我们复现了Alphafold的训练与预测过程,为大家带来第一手的测试体验。
模型介绍
AlphaFold解决的问题是蛋白质折叠问题。输入是一个氨基酸序列,每一个位置代表一个元素,输出是一个拓扑结构,如下图所示:
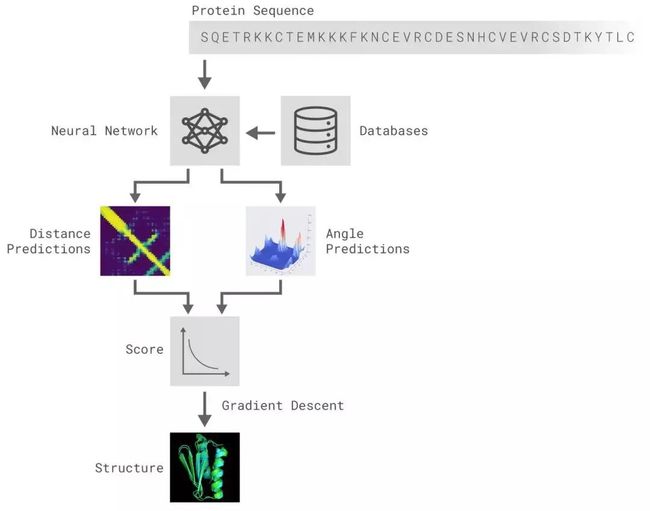
直接输出一个拓扑结构对于深度学习来说比较困难,一般需要输出结构化的数据。DeepMind将该问题转化为预测氨基酸链的一些性质。准确的来说,深度模型输出的是每一个氨基酸单元和其下一个氨基酸单元在空间中的夹角与距离,再组合成拓扑结构。

如上图所示,AlphaFold算法大致分为以下几个部分:
-
特征工程:序列和MSA特征抽取,结合专家经验数据库,把氨基酸链的输入转换到特征空间;
-
深度神经网络结构预测:依据1中的特征预测氨基酸链的一些性质,比如氨基酸之间两两的距离分布,氨基酸链的夹角分布;
-
Potential Construction:结合专家经验构造一个评估函数,来评估2中神经网络输出解的合理程度;
-
结构生成:对于2中预测的距离分布、夹角分布,使用3中的评估函数评估Loss,然后使用梯度下降法优化,直到收敛。
可以看到,Alphafold核心贡献在于:
-
深度学习模型融入专家经验:特征构建阶段,尽可能结合领域知识构建特征,以提高模型表现;评估阶段,将专家知识(氨基酸链的物理特性)结合到损失函数中,进行更精准的判断。
-
全新的蛋白质折叠问题的建模方式:DeepMind不是直接去预测蛋白质的结构,而是将该问题转化为预测氨基酸折叠后的一些性质,再将这些性质转化为可微的约束,并求解。这种思路可以应用在很多复杂的求解问题上。
-
对于梯度下降算法的优化:DeepMind提出通过类似进化/退火算法,解决梯度下降中的局部最优问题,以求解真正的“最优解”。
模型实践
DeepMind仅开源了Alphafold的预测代码和预训练好的模型参数,而对于训练部分,OpenFold与UniFold分别开源了Pytorch和Tensorflow的版本。笔者选择Openfold进行改造,因为其复现完整度比较高,且使用Pytorch框架更适配幻方AI的运行环境。
项目地址:
https://github.com/aqlaboratory/openfold
STEP1 数据集
从上文可以知道,Alphafold数据集包含两个部分:氨基酸序列数据、专家经验数据。具体的,数据说明如下:
-
bfd / small_bfd:Big Fantastic Database,由论文作者 Martin Steinegger 构建的蛋白质序列数据库,包含65983866个蛋白质家族(MSAs)和2204359010个蛋白质序列(HMMs),总计7个文件1.8T数据量。
-
mgnify:一个微生物基因组数据库,使用JackerHammer工具进行检索,总计一个文件64G数据量。
-
pdb70:一个蛋白质结构数据库,总计9个文件56G数据量。
-
pdb_mmcif:一个蛋白质结构数据库,包含超过55000种不同蛋白质的180000多个3D结构,总计约18w个文件206G数据量。
-
uniclust30:1.3亿条氨基酸序列,使用hhsuite中的HHblits工具进行快速msa检索,总计13个文件86G数据量。
-
uniref90:1.3亿条氨基酸序列,是msa检索所需要的库,使用JackerHammer工具进行检索,总计1个文件58G数据量。
对于 pdb_mmcif 数据集,18w个小文件的高频读写会给集群的存储系统造成不小压力。对应的,我们需要将数据转化成FFRecord数据格式,合并多个小文件以减小磁盘IO的开销。FFRecord (FireFlyer Record) 数据格式是幻方AI自研的简单高效的存储二进制记录的文件格式。具体的:
from ffrecord import FileWriterout_file = os.path.join(mmcif_path, "mmcifs.ffr")writer = FileWriter(outfile, 184929)num, mapids = 0, {}for file in os.listdir(os.path.join(mmcif_path, "mmcif_files")):with open(os.path.join(mmcif_path, "mmcif_files", file), "r") as fp:mmcif_string = fp.read()ms = pickle.dumps(mmcif_string)writer.write_one(ms)mapids[file[:-4]] = numnum += 1with open(os.path.join(mmcif_path, "mmcifs_index.pk"), "wb") as fp:pickle.dump(mapids, fp)
读取ffr文件:
from ffrecord import FileReadermmcif_reader = FileReader(os.path.join(mmcif_dir, "mmcifs.ffr"), check_data=True)with open(os.path.join(mmcif_path, "mmcifs_index.pk"), "rb") as fp:mmcif_index = pickle.load(fp)if hit_pdb_code in mmcif_index:idx = [mmcif_index[hit_pdb_code]]data = mmcif_reader.read(idx)cif_string = pickle.loads(data[0])
以上数据已集成进幻方AI的数据集仓库,如果需要利用该数据集进行科研的朋友可以直接拉取。
STEP2 预处理
从上文我们知道,Alphafold需要与专家经验库进行交互,抽取MSA特征。这里用到了多个生物学工具:
-
openmm:用于分析模拟的高性能工具包,是 Omnia(预测生物分子模拟的工具套件)的一部分。
-
kalign:一款针对大规模基因组序列的多序列对比工具,运行速度与准确度都比较高。
-
jackhmmer:一款高效的蛋白质搜索工具,用于从序列数据库中,找到同源的序列。
-
pdbfixer:用于修复PDB数据,对其中蛋白质结构进行基本的准备或者修复。
-
HHsuite:一款蛋白质研究软件,快速搜索分析蛋白质的功能特性。
以上工具都只能使用CPU多核进行加速计算,对CPU的算力比较依赖。对应的,在幻方AI的超算系统结构中,GPU算力池与CPU算力池是分离开的。因此,数据预处理与模型计算要拆分成两个模块单独进行。
name: precompute_alignmentscode:directory: /ceph-jd/prod/jupyter/hwj/notebooks/openfoldentrypoint: scripts/precompute_alignmnets.pyparameters: mmcif/ alignments/ --cpus 250resource:- container: cuda_11- node_count: 1group: jd_dev_alphapriority: 20
这里,jd_dev_alpha 是CPU计算池。通过 hfai run 提交上述 yaml 文件便可以发起对于目标蛋白质结构的预处理任务。
STEP3 模型训练
openfold 采用了Pytorch_lightning来构建整个训练代码,其自动封装了dataloader、多机多卡训练、checkpoint保存与打断自恢复等诸多逻辑,可以直接适配萤火二号的智能分时调度系统。这里我们通过 hfai run 运行下述Yaml文件:
name: precompute_alignmentscode:directory: /ceph-jd/prod/jupyter/hwj/notebooks/openfoldentrypoint: train_openfold.pyparameters: mmcif/ alignments/ data/pdb_mmcif output 2021-10-10 --gpus 1 --resume_from_ckpt output/resume_checkpoint.ckptresource:- container: cuda_11- node_count: 1group: jd_a100priority: 20
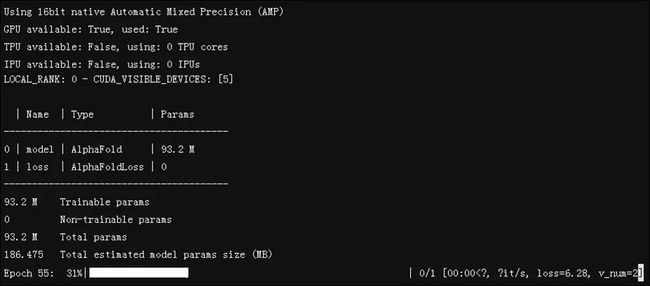
这里,jd_a100 是GPU计算池。由于目标预测的样本不多,这里使用一个计算卡进行训练,过程如下图所示:
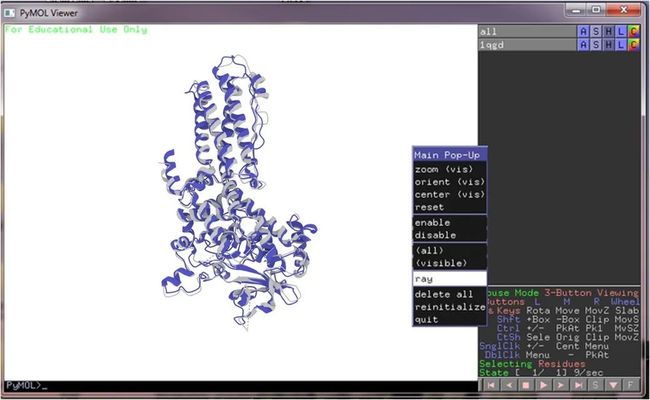
训练结束后,我们加载输出的训练模型,预测蛋白质的结构,得到PDB文件。通过PyMol3d工具,可以将PDB中的蛋白质结构数据可视化出来,最终效果如下:
体验总结
Alphafold作为今年火爆全球的人工智能领域重点突破之一,开启了AI+生物医药领域的新时代。借助萤火二号和自研文件系统,我们可以比较高效地训练该模型。同时,该模型不同于以往深度学习训练场景,其特征抽取与训练,对CPU与GPU的计算需求都比较高。在我们的测试中,GPU整体使用率不高,在20%~30%左右,训练的时间瓶颈是在CPU的计算上。我们欢迎感兴趣的学者们加入我们,继续在幻方萤火二号上对该课题进行进一步探索。
综合体验打分如下:
01:研究新颖度 ★★★★★
该模型开创了AI+生物的新课题,引领了一个新的AI时代。
02:开源指数 ★★★
数据已开源,代码开源了预测部分,训练代码开源需要依赖第三方实现。
03:算力门槛 ★★★★★
数据量大,需要CPU与GPU双重算力支持。
04:通用指数 ★★
模型专注于蛋白质结构预测,有大量的专家经验结合,通用性比较差。
05:模型适配度 ★★★★★
需要CPU与GPU的双重算力,整体GPU利用率不高。同时代码和数据都有一定改造工作。
附录
论文标题:
Improved protein structure prediction using potentials from deep learning
原文地址:
https://www.nature.com/articles/s41586-019-1923-7
源码地址:
https://github.com/aqlaboratory/openfold