2022-LCLR-DIFFDOCK: DIFFUSION STEPS, TWISTS, AND TURNS FOR MOLECULAR DOCKING
2022-LCLR-DIFFDOCK: DIFFUSION STEPS, TWISTS, AND TURNS FOR MOLECULAR DOCKING

Paper: https://arxiv.org/abs/2210.01776
Code: https://github.com/gcorso/DiffDock
预测小分子配体与蛋白质的结合结构(称为分子对接)是药物设计的关键。最近的深度学习方法将对接视为一个回归问题,与传统的基于搜索的方法相比,它减少了运行时间,但在准确性方面还没有得到实质性的改进。相反,将分子对接构建为生成建模问题,并开发了DIFFDOCK,这是配体姿态的非欧几里得流形上的扩散生成模型。为此,将此流形映射到涉及对接的自由度(平移、旋转和扭转)的积空间,并在此空间上开发有效的扩散过程。举例来说,DIFFDOCK在PDB- Bind上获得了38%的top-1成功率(RMSD<2 a),显著优于之前最先进的传统对接(23%)和深度学习(20%)方法。此外,DIFFDOCK具有快速推断时间和提供高选择精度的置信度估计。
蛋白质的生物学功能可以通过与之结合的小分子配体(如药物)来调节。因此,计算药物设计中的一个关键任务是分子对接,预测配体与目标蛋白结合时的位置、方向和构象,从而推断配体的作用(如果有的话)。传统的对接方法[Trott & Olson, 2010;Halgren等人,2004]依赖于估计所提议结构或姿态正确性的评分函数,以及搜索评分函数的全局最大值的优化算法。然而,由于搜索空间很大,评分函数的范围很广,这些方法往往太慢,而且不准确,特别是对于高通量工作流。
近期作品[St ark et al., 2022;Lu等人,2022]开发了深度学习模型,一次性预测绑定姿势,将对接视为回归问题。虽然这些方法比传统的基于搜索的方法快得多,但它们还没有证明在准确性方面有显著提高。,这可能是因为基于回归的范式与分子对接的目标不完全对应,这反映在这样一个事实中,即标准精度指标类似于预测模型下数据的可能性,而不是回归损失。因此,将分子对接构建为给定配体和目标蛋白结构的生成建模问题,学习配体姿势的分布。
因此,作者开发了一种用于分子对接的配体空间上的扩散生成模型(DIFFDOCK)。定义了一个涉及到对接的自由度的扩散过程:配体相对于蛋白质的位置(定位结合袋),它在口袋中的方向,以及描述其构象的扭转角。DIFFDOCK样品姿态通过运行学习到的(反向)扩散过程,该过程迭代地将配体姿态上未知的、有噪声的先验分布转换为学习到的模型分布, r如下图所示
 直观地说,这个过程可以被看作是通过更新它们的平移、旋转和扭转角度来逐步优化随机姿势。
直观地说,这个过程可以被看作是通过更新它们的平移、旋转和扭转角度来逐步优化随机姿势。
虽然DGMs已经应用于分子机器学习中的其他问题,但现有的方法不适用于分子对接,其中配体的空间是一个(m+6)维的子流形 M ∈ R 3 n M \in R^{3n} M∈R3n,其中n和M分别是原子数和扭转角。为了开发DIFFDOCK,对接自由度将M定义为通过一组允许的配体位姿转换可访问的位姿空间。使用这种思想将M中的元素映射到与这些转换相对应的组的积空间,在其中可以有效地开发和训练DGMs。由于对接模型的应用通常只需要固定数量的预测和这些预测的置信度分数,训练一个置信度模型,为从DGM中采样的姿态提供置信度估计,并挑选出最有可能的样本。
这个两步过程可以被视为蛮力搜索和一次性预测之间的一种中间方法:保留了考虑和比较多个姿势的能力,而不会遇到高维搜索的困难。
根据经验,在标准盲对接基准PDBBind上,在配体均方根距离(RMSD)低于2 A的情况下,DIFFDOCK实现了38%的top-1预测,几乎是之前最先进的深度学习模型(20%)的两倍。DIFFDOCK的性能甚至超过了最先进的基于搜索的方法(23%),同时在GPU上仍然快3到12倍。此外,它提供了其预测的准确置信度评分,在其最自信的三分之一之前未见的复合体上获得83% RMSD<2 A。
综上所述,本工作的主要贡献是:
- 将分子对接任务构建为一个生成问题,并强调了之前深度学习方法的问题。
- 提出了一个新的扩散过程的配体姿态对应的自由度涉及分子对接
- 在PDBBind盲对接基准上, R M S D < 2 A RMSD<2 A RMSD<2A,实现了最新的38%的top-1预测,大大超过了之前最好的基于搜索的方法(23%)和深度学习方法(20%)。
Molecular docking. 分子对接任务通常分为已知口袋对接和盲对接。已知口袋对接算法将分子在蛋白质上结合的位置(结合口袋)作为输入,只需要找到正确的方向和构型。由于大多数情况下蛋白质的相对刚性,对接方法通常假设已知结合的蛋白质结构,将遵循这一假设。方法通常通过命中百分比或近似正确的预测来评估,通常认为配体RMSD误差低于2 A [Alhossary等人,2015;哈桑等人,2017;麦克纳特等人,2021年]。
Search-based docking methods. 传统对接方法[Trott & Olson, 2010;Halgren等人,2004年;Thomsen和Christensen, 2006]由一个参数化的基于物理的评分函数和一个搜索算法组成。评分函数采用3D结构并返回给定姿态的可能性估计值,而搜索则随机修改配体姿态(位置、方向和扭转角),目标是找到评分函数的全局最优值。这些基于搜索的方法在对接到一个已知的口袋时提供了相对的改进,但运行起来通常计算成本非常高,而且必须努力解决盲对接所特有的非常大的搜索空间。
Machine learning for blind docking. 最近,EquiBind [St ark et al., 2022]试图通过直接预测配体和蛋白质上的口袋关键点并对准它们来解决盲对接任务。TANKBind [Lu等人,2022]通过独立预测每个可能的口袋的对接姿势(以原子间距离矩阵的形式),然后对它们进行排序,改进了这一点。虽然这些基于单次或少次回归的预测方法速度快了几个数量级,但其性能还没有达到传统的基于搜索的方法。
Diffusion generative models. 设数据分布为连续扩散过程的初始分布 p 0 ( x ) p_0(x) p0(x),描述为 d x = f ( x , t ) d t + g ( t ) d w dx = f (x,t) dt + g(t) dw dx=f(x,t)dt+g(t)dw,其中 w w w为Wiener过程。扩散生成模型(DGMs)对扩散数据分布的score ▽ x l o g p t ( x ) \bigtriangledown x log p_t(x) ▽xlogpt(x)进行建模,以便通过反向扩散生成数据 d x = [ f ( x , t ) g ( t ) 2 x l o g p t ( x ) ] + g ( t ) d w dx = [f (x,t) g(t)2 x log p_t(x)] + g(t) dw dx=[f(x,t)g(t)2xlogpt(x)]+g(t)dw [Song等人,2021]。在这里,总是令 f ( x , t ) = 0 f (x,t) = 0 f(x,t)=0。已经为分子机器学习任务开发了几种DGMs,包括分子生成[Hoogeboom等人,2022]、构象生成[Xu等人,2021]和蛋白质设计[Trippe等人,2022]。然而,这些方法学习了在整个欧氏空间 R 3 n R^{3n} R3n上的分布,每个原子有3个坐标,这使得它们不适用于自由度受到更大限制的分子对接。
model
尽管EquiBind和其他ML方法通过避免昂贵的配体姿态优化过程提供了强大的运行时改进,但它们的性能还没有达到基于搜索的方法。正如下面的分析所指出的,这可能是由模型的不确定性和目标函数的优化所引起的,该目标函数与分子对接在实践中如何使用和评估并不对应。
Molecular docking objective 分子对接在药物发现中起着至关重要的作用,因为预测结合蛋白配体复合物的3D结构可以进一步对结合相互作用的强度和性质进行计算和人类专家分析。因此,对接预测只有在其与真实结构的偏差不显著影响此类分析的输出时才有用。因此,停靠预测只有在其与真实结构的偏差不显著影响此类分析的输出时才有用。因此,在该领域中使用的标准评价度量一直是配体RMSD(晶体配体位姿)低于某个值 ϵ \epsilon ϵ的预测的百分比。
然而,在某些容忍范围内,最大化RMSD预测的比例的目标是不可微的,不能用于随机梯度下降的训练。然而,在某些容差 ϵ \epsilon ϵ内最大化RMSD预测的比例的目标是不可微的,不能用于随机梯度下降的训练。相反,当RMSD < ϵ \epsilon ϵ时,最大化预测的预期比例对应于在模型的输出分布下,在 ϵ \epsilon ϵ趋于0的极限下,最大化真实结构的可能性。这一观察促使训练生成模型,以最小化模型分布下所观察结构的负对数似然的上界。因此,将分子对接视为在蛋白质结构上学习分布的问题,并在该空间上开发了扩散生成模型(第4节)
Confidence model 使用训练过的扩散模型,可以根据模型从后验分布中采样任意数量的配体位姿。然而,研究人员通常只对一个或少量的预测姿态感兴趣,并对下游分析的相关置信度进行测量。因此,在扩散模型采样的姿势上训练一个置信度模型,并根据它们在容错范围内的置信度对它们进行排序。然后将排名第一的配体姿态和相关置信度作为DIFFDOCK的排名第一的预测和置信度得分.
**Problem with regression-based methods. **. 开发用于分子对接的深度学习模型的难点在于姿态的数据固有(任意)不确定性(多个姿态可能是正确的),以及与有限的模型容量和可用数据相比任务的复杂性(认知不确定性)。因此,考虑到现有的协变量信息(只有蛋白质结构和配体身份),任何方法都将在许多可行的替代方案中对正确的结合姿势表现出不确定性。任何回归风格的方法,被迫选择一个单一的配置,使期望的平方误差最小化,将学会预测(加权)这些替代的平均值。相比之下,具有相同协变量信息的生成模型将旨在捕获备选方案的分布,填充大多数重要模式,即使同样无法区分正确的目标。如下图所示,这种行为导致基于回归的模型比作者的方法产生明显更多的物理上不可信的姿势。

特别是,观察到频繁的空间冲突(例如,在EquiBind的预测中占26%)以及EquiBind和TANKBind的预测中的自交叉(图5和图9)。

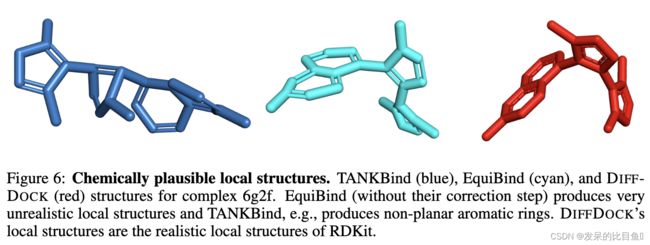
在DIFFDOCK的预测中没有发现交集。这些现象的可视化和定量证据见附录E.1。
配体位姿是 R 3 R^3 R3中原子位置的分配,因此原则上,可以将位姿x视为 R 3 n R^{3n} R3n中的一个元素,其中n是原子数。然而,这包含了比分子对接相关的更多自由度。特别是,配体中的键长、角度和小环基本上是刚性的,因此配体的灵活性几乎完全取决于可旋转键的扭转角度。传统的对接方法,以及大多数的ML方法,都以隔离配体的种子构象 c ∈ R 3 n c \in R^{3n} c∈R3n作为输入,仅改变最终束缚构象的相对位置和扭转自由度。因此,与 c c c一致的配体位姿空间是一个 ( m + 6 ) (m + 6) (m+6)维子流 M c R 3 n Mc R3n McR3n,其中 m m m是可旋转键的数量,另外六个自由度来自于相对于固定蛋白质的旋转平移。遵循这样的范式,将种子构象 c c c作为输入,并将分子对接制定为学习基于蛋白质结构 y y y的歧管 M c M_c Mc上的概率分布 p c ( x ∣ y ) p_c(x |y) pc(x∣y)。
De Bortoli等人[2022]通过将环境空间中的扩散投影到子流形上,制定了子流形上的 D G M s DGMs DGMs。然而,这种扩散的核 p ( x t ∣ x 0 ) p(x_t | x_0) p(xt∣x0)不能以封闭形式获得,必须用测地线随机游走进行数值采样,这使得训练效率非常低。相反,定义了一个一对一映射到另一个更好的流形,其中扩散核可以直接采样,并在该流形中开发一个 D G M DGM DGM。首先,重申最后一段中的讨论如下:

这可以看作是流形 M c M_c Mc的非正式定义。同时,给出了一个连续的与 m + 6 m + 6 m+6自由度相对应的配体位姿变换族,可以将 M c M_c Mc上的一个分布提升为相应群的积空间上的一个分布,这个分布本身就是一个流形。然后,将展示如何在这个积空间上对扩散核进行采样,并在上面训练 D G M DGM DGM。
定义 S O ( 2 ) m SO(2)^m SO(2)m元素的操作,使其对结构造成最小扰动(RMSD意义上)

这些属性可以更正式地表述如下(证明在附录A中)

接下来展示如何使用积空间来学习 M c M_c Mc中配体位姿上的DGM。理论结果(证明在附录a中)。

SO(3)上的扩散核由IGSO(3)分布给出[Nikolayev & Savyolov, 1970;Leach等人,2022],可以在轴-角参数化中通过采样单位矢量 ω ^ ∈ s o ( 3 ) \hat{ω} \in so(3) ω^∈so(3)均匀7和随机角度 ω ∈ [ 0 , π ] ω \in [0,π] ω∈[0,π]根据进行采样
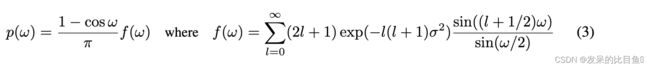
Diffusion model. 为评分模型提供完整的3D结构,而不是产品空间的抽象元素,使其能够使用SE(3)等变模型推理物理相互作用,而不依赖于扭转角的任意定义,并更好地推广到未见的复合体。
Confidence model. 为了收集置信度模型d(x,y)的训练数据,运行训练过的扩散模型,为每个训练示例获得一组候选姿态,并通过测试每个姿态的RMSD是否低于2A来生成标签。
实验

Docking accuracy. DIFFDOCK显著优于之前的所有方法(表1)。特别是,在采样40个姿势时,DIFFDOCK获得了令人印象深刻的38.2%的top-1成功率(即RMSD <2 A8的预测百分比),而在采样10个姿势时,则获得了35.0%。这一性能大大超过了最先进的商业软件,如GLIDE(21.8%)和先前最先进的深度学习方法TANKBind(20.4%)。基于ML的口袋预测与基于搜索的对接方法相结合的使用提高了基线性能,但即使是其中最好的(P2Rank+GNNA)也仅达到28.8%的成功率。
Inference runtime. DIFFDOCK拥有其卓越的准确性,(在GPU上)比最好的基于搜索的方法GNINA快3到12倍(表1)。这种高速度对于应用至关重要,例如对候选药物的高通量虚拟筛选或对蛋白质靶点的反向筛选,在这些应用中,人们经常搜索大量的复合物。

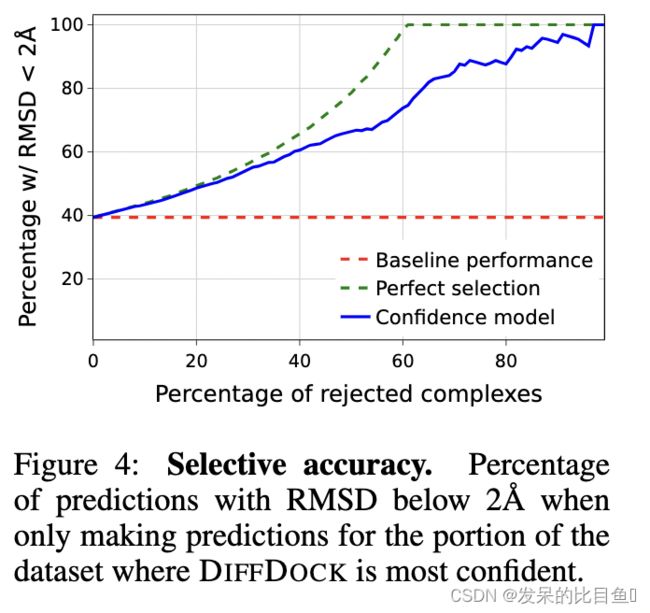
Selective accuracy of confidence score. 正如前1的结果所示,DIFFDOCK的置信度模型在对给定复合体的采样姿势进行排名并选择最佳姿势时非常准确。通过评估DIFFDOCK仅在置信度高于某一阈值(称为选择性预测)时进行预测,来研究不同复合物的置信度模型的选择准确性。在图4中,绘所做预测的复合体的百分比时的成功率,即增加置信度阈值。