论文解读《STALLION:一个基于堆叠的原核赖氨酸乙酰化位点预测的集成学习框架》
期刊:Briefings in Bioinformatics
分区:二区
摘要
蛋白质翻译后修饰(PTM)是一种重要的调节机制,在正常和疾病状态中都发挥着关键作用。赖氨酸残基上的乙酰化是由于其在细胞代谢和调节过程中的重要作用之一。识别蛋白质赖氨酸乙酰化(Kace)位点是生物信息学中的一项具有挑战性的任务。到目前为止,几种基于机器学习的方法,在硅中识别Kace站点的方法已经被开发出来。其中,少数是原核生物物种特异性的。尽管这些方法具有诱人的优点和性能,但它们也有一定的局限性。因此,本研究提出了一种新的预测STALLION(基于堆叠的原核赖氨酸氨基酸预测器),包含6个原核物种特异性模型,以准确识别Kace位点。为了提取Kace站点周围的关键模式,我们使用了11种不同的编码,代表了三种不同的特征。随后,采用系统、严格的特征选择方法,独立识别五种基于树的集成算法的最优特征集,并为每个物种建立各自的基线模型。最后,利用基线模型的预测值,利用适当的分类器训练来发展STALLION。比较基准实验表明,STALLION在独立测试中显著优于现有的预测器。为了加快对STALLION模型的直接访问,实现了一个用户友好的在线预测器,可在:http://thegleelab.org/STALLION上获得。
引言
分子生物学的“中心信条”的最后一步是翻译过程,其中RNA编码特定的蛋白质。蛋白质翻译后修饰(PTMs)具有重要意义,因为它们在一些生物过程中发挥了作用,包括细胞周期调节、DNA修复、基因激活、基因调控和信号转导过程。是蛋白质生物合成后期发生的可逆或不可逆化学变化。PTMs可以发生在单个氨基酸残基或多个残基中,导致改变位点的化学性质的改变。可逆修饰包括共价修饰,而不可逆变化包括蛋白水解修饰。PTMs可以影响蛋白质的多种特性,如细胞-细胞/细胞-基质相互作用、酶组装和功能、分子运输、蛋白质-蛋白质相互作用(PPIs)、蛋白质折叠、蛋白质定位、蛋白质溶解度、蛋白质寿命和受体激活,从而作为蛋白质功能的重要调节工具。超过400种不同类型的经前磁刺激已被鉴定,从添加小的化学或复杂基团(即。乙酰化、甲基化、磷酸化等)。以及多肽的添加。泛素化和sumo化)。赖氨酸残基经历了最多的PTMs与至少15个PTM类型。根据dbPTM数据库的统计,磷酸化、乙酰化和泛素化是覆盖>90%已报道的的三种主要类型.
赖氨酸乙酰化(Kace)是最重要的普遍存在的物质之一,在原核生物和真核生物中都高度保守。这是一个共价PTM催化赖氨酸乙酰转移酶(KATs),乙酰基(CH3CO)从乙酰辅酶转移到自由α氨基(氨+)的n端残基(Nα乙酰化)或ε氨基内部赖氨酸(Nε乙酰化)在特定的网站。乙酰化有三种类型。Nα-Nε和O-乙酰化。Nε-和o-乙酰化是可逆的修饰,而Nα-乙酰化是不可逆的。Nα-乙酰化在真核生物中很常见,而Nε-乙酰化在生物学上更为重要,在肌动蛋白成核、细胞周期调控、染色质稳定性、细胞代谢、核运输和PPIs中发挥重要作用。Kace的失调也与衰老和一些疾病有关,包括癌症、免疫疾病和心血管和神经系统疾病。鉴于乙酰化在细胞生物学和疾病病理中很重要,识别Kace位点对于理解其调节机制是必要的。
近年来,一些实验方法,包括放射性化学方法、质谱和染色质免疫沉淀,已被开发用于检测Kace PTM位点。由于科学技术的最新创新,我们检测Kace位点的能力大大提高;然而,考虑到蛋白质组的大小,我们只发现了一点点赖氨酸“修饰组”。此外,测试单一蛋白质中的每一个赖氨酸残基都是很费力的。复杂的实验识别凯网站(耗时、昂贵、劳动密集型和低吞吐量)导致过多的计算方法设计预测潜在的实验验证,特别是机器学习(ML)工具,已经变得越来越流行的快速和准确的预测。在过去的十年里,一些ML技术已经被使用开发用于鉴定原核生物和真核生物中的Kace位点。
目前,有十几种Kace预测工具可用,例如PAIL , LysAcet , EnsemblePail , N-Ace , BPBPHKA , PLMLA , PSKAcePred , KAcePred , LAceP , AceK , SSPKA , iPTM-mLys , KA-predictor , ProAcePred , ProAcePred 2.0 , Ning et al. and DNNAce。大多数预测因子被设计用于鉴定真核生物中的乙酰化,且缺乏物种特异性。然而,有一些现有的预测因子已经被开发出来用于识别原核生物中的Kace。SSPKA和ka预测因子被开发用于真核和原核乙酰化位点的预测,其中包括两个原核生物,,从而强调了一个物种特异性模型的重要性和必要性。Chen等人[36]开发了一种名为ProAcePred的预测9种原核生物、古菌、枯草芽孢杆菌、谷氨酸棒状杆菌、大肠杆菌、嗜地杆菌、结核分枝杆菌、鼠伤寒杆菌和副溶血弧菌。后来,同一组为6种原核生物开发了更新版本的ProAcePred预测器ProAcePred 2.0: B.subtilis C. 谷氨酰胺、大肠杆菌、杆菌、结核分枝杆菌和鼠伤寒沙门氏杆菌。训练数据集略大于ProAcePred中使用的数据集。这种ML研究为了解原核生物和真核生物之间底物位点特异性的差异提供了机会。
虽然在Kace站点的计算预测方面已经取得了进展,但仍有一些局限性需要解决。首先,大多数最先进的方法都使用了简单的ML算法,如支持向量机(SVM)或随机森林(RF)来训练模型。由于尖端技术的进步,先进的ML方法,如深度学习(DL)、迭代特征表示或基于集成的堆叠方法,可以用来开发一个更鲁棒和稳定的预测器,以提高Kace站点的预测性能。其次,现有方法在Kace预测中所使用的特征空间相当有限。最后,最先进的方法使用简单的特征选择技术来识别最优特征子集。不幸的是,这种简单的方法可能会忽略Kace站点预测中存在的关键特征。考虑到这些局限性,我们开发了一种新的基于堆叠的预测器,称为STALLION(基于堆叠的原核赖氨酸堆叠预测器),以提高对6个不同原核生物物种的Kace位点的准确预测。与其他最先进的方法相比,我们提出的方法的主要优点如下:(i)STALLION是第一个基于堆叠集成的预测原核生物Kace位点的预测器;(ii)我们对每个物种的综合评估和比较了每个物种的11种不同的编码方案,并试图提取代表广泛序列、位置特异性和物理化学特征的模式。随后,我们使用三种不同的计算密集型方法分别对五种流行的基于树的集成算法确定最优特征集,并对基分类器进行训练。(iii)使用来自基分类器的预测信息和5倍交叉验证,使用适当的分类器训练叠加模型STALLION。对独立数据集的比较分析显示,该STALLION显著优于现有的预测器,从而突出了利用我们的系统方法进行Kace预测的重要性。
数据和方法
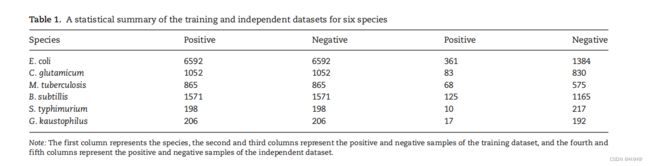
训练和独立的数据集
最近,Chen等人基于PLMD数据库http://plmd.biocuckoo.org/,为6个物种构建了新的非冗余数据集, (B. subtilis, C. glutamicum, E. coli, G. kaustophilus and M. tuberculosis)。因此,采用CD-HIT通过将序列同一性阈值设置为30%来消除同源序列,这对于避免交叉验证或模型训练中的高估具有很大的价值。在构建数据集时,作者使用不同的片段大小进行实验,确定了最优大小为21个残基长的序列片段,中心为K。如果中心K残基乙酰化经过实验验证,则这些片段被定义为阳性样本(Kace),否则它们就被认为是阴性(非Kace)样本。值得注意的是,缺少残基的中心K或任意末端的间隙被一个虚拟原子“O”取代。利用这些数据集,他们开发了一种名为ProAcePred2.0的物种特异性Kace位点预测器。
我们在当前的研究中使用了相同的数据集,因为它们是最近构建的,并使用了严格的方法来确定最优长度。一般来说,使用这样一个高质量的数据集开发一个预测模型可能有更全面的实际应用。每个物种的训练和独立数据集的统计汇总如表1所示。我们使用平衡训练数据集进行预测模型开发,使用不平衡独立数据集来检验模型的鲁棒性。
特征编码方案的选择
为了建立一种有效的基于ML的Kace预测方法,我们采用了几种不同的特征编码方案来编码21种氨基酸[20个标准氨基酸和gap(O)的虚拟残基]。我们总共采用了11种编码方案,可分为三种主要类型:(i)基于序列的特征包括氨基酸(NRA)、二进制编码(BINA)、氨基酸组成(AAC)、二肽组成(DPC)和联合三联体(CTF);(ii)基于物理化学性质的特征包括氨基酸指数(AAI)、分组二肽组成(GDPC)、分组三肽组成(GTPC)、k间隔氨基酸组对组成(CKSAAGP)和Zscale,(iii)位置特异性评分矩阵包括BLOSUM62。
特定于位置的评分矩阵
BLOSUM62 (BLOS).BLOSUM62矩阵通常应用于爆炸序列比对程序中。在这里,它被用来转换蛋白质序列来描述两个序列片段的相似性。一般来下,该替代矩阵用于大型数据库中相关蛋白的序列保守性研究,已被用作多个预测因子的特征。BLOSUM62矩阵中的每一行都可以用来编码这20个氨基酸中的一个。因此,我们可以根据BLOSUM62矩阵进行编码,形成一个420(20×21)D的特征向量。
ML算法的选择
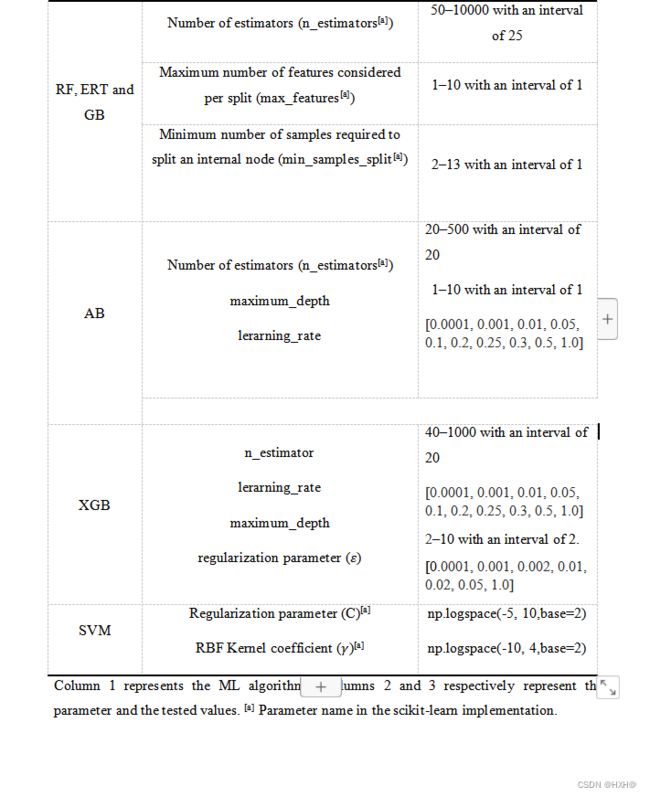
在本研究中,我们采用了6种不同的分类器,包括5种基于决策树的分类器(RF、极端梯度增强算法(XGB)、AdaBoost(AB)、梯度增强(GB)、极随机树(ERT)和SVM。一般来说,基于决策树的算法可以处理非标准化的特征,而不像其他监督和DL算法。因此,我们只使用这5个分类器来构建基线模型。然而,我们使用了6个分类器来构建元模型,并选择了一个合适的分类器。这些分类器已经在计算生物学和生物信息学中得到了许多成功的应用。每个分类器的详细过程与我们之前的研究一致。通常,需要k倍交叉验证分析来训练或开发预测模型。我们采用了5倍交叉验证,并使用网格搜索方法确定了最优超参数。补充表S1提供了每个分类器的网格搜索空间。
STALLION的框架
图1总结了一个基于叠加集成学习的种马框架。它涉及整体工作低的三个关键步骤,描述如下:

特征表示
每个物种训练数据集中的序列基于AAI(525D)、AAC(21D)、DPC(441D)、cksaagp(150D)、CTF(343D)、Zscale(101D)、BINA(441D)、Blos(420D)、GTPC(125D)、GDPC(25D)和NRF(20D)编码方案进行编码。我们对每个序列的这11个编码进行线性积分,得到一个2616D特征向量。因此,对的训练数据集进行了分析 B. 精细的 C. 谷氨酰胺、大肠杆菌、考斯托夫杆菌、结核分枝杆菌和鼠伤寒杆菌分别为3142×2616、2104×2616、13、184×2616、412×2616、1730×2616和396×2616矩阵。
功能优化与选择
每个序列都包含一个高维的特征向量(2616D),它可能包含不相关的或冗余的信息。因此,在模型训练过程中,预测性能下降,需要大量的计算资源。我们采用两步特征选择策略,从原始特征维中选择信息最丰富的特征。在第一步中,每个特性都会根据评分函数得到一个分数。在这里,我们使用了三种不同的评分函数,即。F-score、RF(RFIS)估计的特征重要性评分(FIS)和XGB(XFIS)计算的FIS,根据它们区分Kace和非Kace站点的能力。然后,我们根据其得分对原始特征维度进行降序排序。我们总共生成了三个特征列表(F-score,RFIS和XFIS),其中F-score和RFIS包含前2000个特征,而XFIS包含只有非零值的特征(∼500个特征)。
其次,在对三个特征列表上独立应用序列前向搜索(SFS)来识别次优特征子集。字母r和字母s分别表示已排序的特征列表和次优子集。在SFS中,k(k=5表示F-score和RFIS;k=2表示XFIS)将信息最丰富的特征从r移动到s,分别输入到5个不同的分类器中,并通过在s中采用5倍交叉验证来评估性能。这个过程被重复,直到r变空。最终,每个具有优越的Mathews相关系数(MCC)性能的分类器的特征子集被认为是每个物种的最优集。通常,将使用其中一个评分函数和一个分类器来确定最优特征集。然而,我们应用了一个系统的方法来识别最优特征集,尽管这个过程是计算广泛的。由于我们使用了3个不同的排序列表和5个不同的分类器,我们获得了每个物种的15个模型。
堆叠集成学习
对于每个分类器,我们从三个不同的次优子集模型中选择最佳模型。因此,我们获得了每个物种的5个最优基线模型。将从基线模型中接收到的预测概率和类别标签结合起来,并作为一个新的特征向量(10D)。一般来说,基线模型的乘积是用逻辑回归训练的,同时开发最终的预测模型。然而,我们探索了6个分类器,其中包括5个基于树的分类器和SVM。包含SVM的原因是新的特征向量在0-1的范围内,SVM可以很好地处理。所有这些分类器都采用了10个随机的5倍交叉验证程序进行训练。鉴于MCC是我们在5倍交叉验证过程中的目标函数,可能有可能过拟合预测模型以达到最高的MCC。因此,我们通过随机划分训练数据集,重复5次5倍交叉验证,为每个分类器得到10个最优特征集。例如,C和γ参数的SVM各有10个值。但是,我们选择了C和γ的中位数参数来建立最终的预测模型。这种随机交叉验证技术可以避免的过拟合。最后,比较从随机的5倍交叉验证中获得的平均性能,以选择每个物种的最佳模型。
附加功能编码
在本研究中,我们还测试了k-最近邻(KNN)的编码。KNN编码根据一个给定序列与来自训练数据集(KAce和非KAce)的n个样本的相似性,对一个给定序列进行特征分析。特别是,对于给定的两个固定长度的序列r1和r2,相似度得分F(R1,R2)计算如下:
其中r1和r2为第j位两个序列的氨基酸残基,K为序列长度。两个氨基酸m和n,相似度评分定义如下: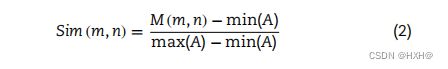
式中(m,n)由BLOSUM62替代矩阵得到的两个氨基酸的相似性得分,A为替代矩阵,min(A)/max(A)分别为矩阵中最小/最大值。在本研究中,我们设置X=2、4、8、16、32、64和128,为给定的序列生成一个7D特征向量。
实施
所有的交叉验证和独立的评估都是在一个使用CentOS Linux 7.6和Python2.7.5的服务器上执行的。值得注意的是,所有的ML分类器(RF、ERT、GB、ERT和XGB ;是由 Scikit-learnv0.18.1软件包构建和优化的。我们计算了三个不同的(F-score,RFIS和XFIS)评分函数,使用相同的包对特征进行排序。此外,本研究中使用的特征编码是使用我们的内部代码进行计算。值得注意的是,一些开源包,如iLearn和iFeftare可以计算这里使用的大多数特征编码。
性能评估策略
采用六种性能测量方法来评估其他研究中广泛使用的模型性能,包括MCC、敏感性(Sn)、特异性(Sp)、准确性(ACC)、平衡准确性(BACC)和受试者工作特征(ROC)曲线下面积(AUC)。这些指标的定义如下:

其中,TP、TN、FP和FN分别表示真阳性、真阴性、假阳性和假阴性。此外,我们还使用ROC曲线和AUC值来评估整体表现。
结果与讨论
不同特征编码方法与分类器之间的性能评估
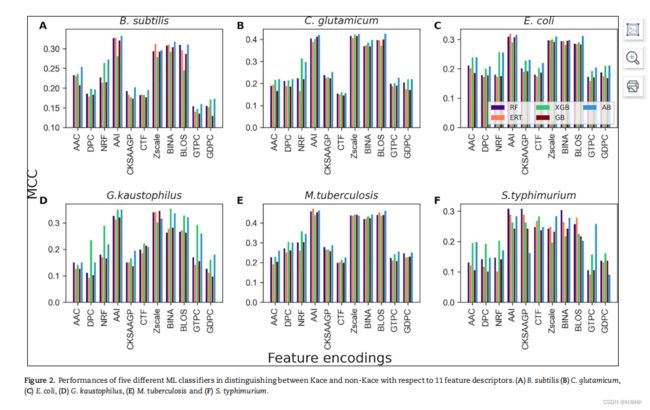
我们采用5种基于树的集成分类器(RF、GB、ERT、XGB和AB)和11种基于序列的特征编码、物理化学性质和位置特异性评分矩阵,系统地研究了各种特征编码和分类器在原核Kace位点预测中的影响。我们对每个物种数据集的每个模型进行了10次随机5倍交叉验证测试,并比较了55个模型(11个编码×5分类器)的性能。图2显示,4种编码(AAI、Zscale、BINA和BLOS)取得了相似的性能,对大多数原核物种的性能显著优于其他7种编码 (B. subtilis, C.glutamicum, E. coli, G. kaustophilus and M. tuberculosis).然而,我们发现6种编码对S. typhimurium的性能相似,且显著高于其他5种编码(AAC、DPC、NRF、GTPC和GDPC)。总的来说,4种编码(AAI、Zscale、BINA和BLOS)均优于同类编码。.然而,其他编码也拥有支持Kace位点预测的必要信息。为了概述每个分类器在Kace预测上的性能,我们计算了每个分类器的66个模型(11个编码×6个物种)的平均性能。结果表明,AB、XGB、RF、ERT和GB的平均MCCs分别为0.261、0.255、0.241、0.232和0.230。值得注意的是,所有的分类器在Kace站点预测中都表现得相当好;然而,AB被发现略微优越。我们可以整合上述信息并开发一个稳健的模型,而不是寻找最好的模型。在本研究中,我们采用了类似于最近研究的堆叠方法。
确定6个物种的每个分类器的最优模型
正如方法部分提到的,我们应用了三种不同的评分函数对特征进行排序,每个都有自己的优缺点。例如,F-score和RFIS为所有给定的特征分配一个相对分数。然而,XFIS排除了∼70%的功能和指定其余特性的相对分数。补充图S1显示了五种分类器对谷氨酰胺种类不同特征集的性能。在这里,我们观察到,基于f-score(补充图S1A)和RFIS(补充图S1B),性能稳步提高,达到了最大的精度,随后保持在平衡状态。然而,对于XFIS,性能增长缓慢,直到最优的一个,随后在添加更多特征的同时恶化(补充图S1C)。

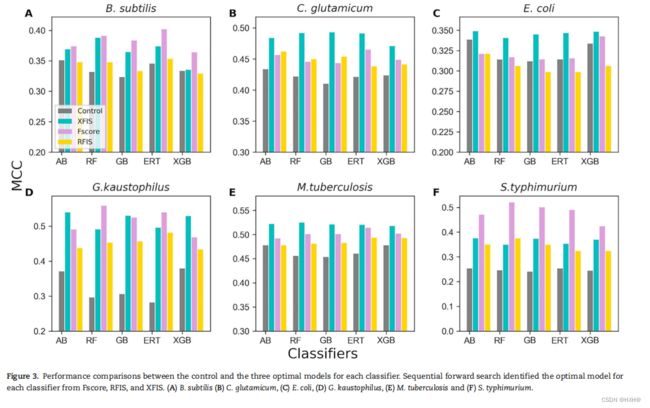
三种不同方法(F-score、RFIS和XFIS)的5个分类器的最优特征集的大小都有所不同。例如,RF、ERT、GB、XGB和AB分别从SFS识别的F-score中获得了1000、520、790、260和410个最优特征集。相应的分类器分别有来自RFIS的140、1290、211、120和150D最优特征集和来自XFIS的30、38、31、52和44D最优特征集。同样,来自三种不同方法的每个分类器的最佳模型显示出不同大小的最优特征集。例如,RF拥有具有1000、140和40D最优特征集的三个模型。然而,我们基于最大的精度选择了最佳的模型。对其他物种采用相同的程序,为每个分类器选择最好的三个模型,并将其性能与对照组进行比较。
从图3中可以看出,最优模型的性能始终优于控制模型,这说明需要采用特征选择技术从原始维度中排除不相关的信息。对于三种植物 (C. glutamicum, E. coli and M. tuberculosis) XFIS得到的最优特征集在5个分类器上具有优于同类分类器(F-score和RFIS)的性能。在两个物种中 (S. typhimurium and B. subtilis)从F-score中提取的最优特征集比(XFIS和RFIS)都取得了良好的性能。然而,对于G. kaustophilus,从F-score得到的最优特征集对RF和ERT分类器的性能有所提高。其余三种分类器在从XFIS获得最优特征后表现出更好的性能。出乎意料的是,来自RFIS的最优特征集并没有表现出最好的性能。值得注意的是,五种分类器的最佳模型被认为是每个物种的基线模型,并用于后续分析。总的来说,我们的系统特征选择分析表明,必须应用不同的评分函数来对特征进行排序,并对SFS分别使用不同的分类器来获得相应的最优特征集。
STALLION的构建
堆叠是一种集成技术,它考虑了不同的预测模型来生成一个稳定的堆叠模型。该方法采用了一种有效的方案来降低各种预测模型的泛化错误率。将5个基线模型的预测值(Kace和类标签的预测概率)相结合,生成一个10D特征向量。与之前的方法不同,我们通过使用10个随机的5倍交叉验证,使用一个新的10D特征向量进行训练,系统地评估了6个不同的分类器(图4)。结果表明,5种分类器(RF、ERT、AB、XGB和SVM)的性能相似,略优于GB。在这五种分类器中,我们选择了三个物种(B. subtilis, C. glutamicum andG. kaustophilus)的AB分类器,针对两个物种的SVM分类器(M. tuberculosis
and S. typhimurium),以及针对E. coli的XGB分类器,其性能略优于其同类产品。6种模型一般命名为STALLION,B. subtilis的ACC、MCC和AUC分别为0.403、0.0700和0.745; C. glutamicum分别为; 0.357, 0.678 和0.733,G. kaustophilus分别为; 0.603, 0.801 和 0.836,M. tuberculosis分别为 0.557, 0.779和 0.782,S. typhimurium.的含量分别为0.571、0.785和0.770。
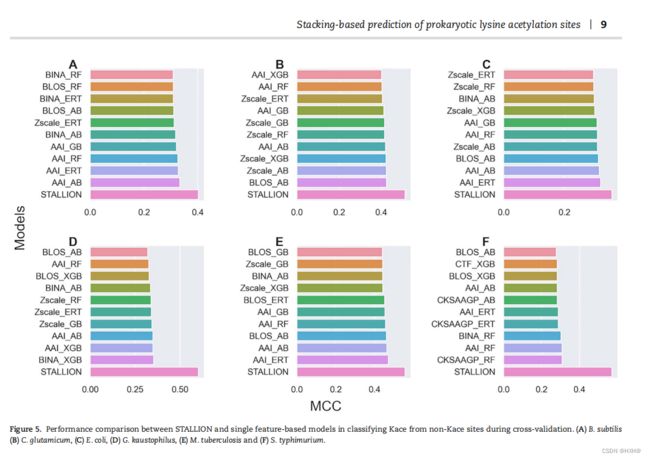
STALLION与单一特征模型的比较
为了展示我们提出的堆叠方法的优势,我们将STALLION与基于单一特征的模型进行了比较。我们从图2中选择了前10个基于单一特征的模型,并将其与6个种物种的 STALLION进行了比较。图5显示,所有6种 STALLION均显著优于单一特征模型,MCC高6.9-9.4% B. 枯草芽孢杆菌属植物,则高出8.8-11.1% C. 谷氨酰胺,大肠杆菌高3.7-6.1%,肠杆菌24.9–28.2%高,结核分枝杆菌高8.6-11.7%,鼠伤寒杆菌高26.2–29.3%。 STALLION的优越性能在单一的基于特征的模型主要是由于新奇的引入我们的方法,其中包括(i)特征融合策略,(ii)从混合特性选择最优特征集为每个分类器独立和各自的基线模型建设和(iii)选择一个合适的分类器叠加模型建设。
特征贡献分析
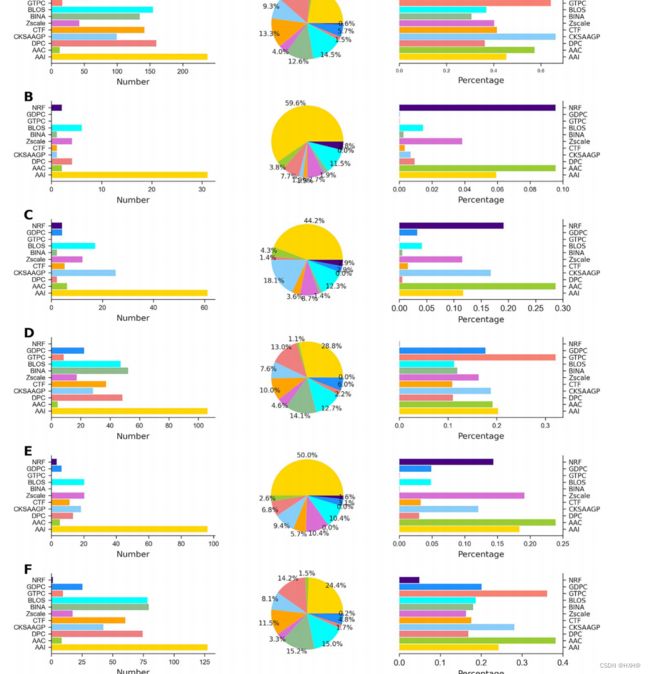
为了了解不同特征在每个物种的最优特征集中的贡献,我们分析了它们的组成和分布。值得一提的是,5个分类器模型对每个物种具有不同的最优特征子集。我们没有关注每个子集,而是考虑了最优特征子集的最大大小,该子集可能包括5个物种的其他四个子集 (B. subtilis, C. glutamicum,
E. coli, M. tuberculosis and S. typhimurium). 在C.glutamicum中,RF、ERT、GB、XGB和AB分别包含30、38、31、52和44D的最优特征子集。在这里,52D有其他的特征子集。然而,在G. kaustophilus中,结合不同的最优亚群来研究它们的作用。从图6可以看出,6个物种间最优特征集的特征分布存在显著差异,但也有一些细微的相似之处。其中,AAI分别占了总最优特征的22.4%、59.6%、44.2%、28.8%、50.0%和24.4% B. subtilis, C. glutamicum, E. coli, G.kaustophilus, M. tuberculosis and S. typhimurium,。这一结果表明,AAI特征对6个物种具有重要的贡献,表明它们在Kace预测中具有重要意义。6个编码序列(AAC、DPC、cksaagp、CTF、Zscale和BLOS)对所有物种的最优特征集均有贡献。尽管如此,它们之间的贡献水平仍然不同,这表明在Kace预测中发挥了支持作用。此外,我们观察到GTPC和GDPC、GTPC、NRF和GTPC和BINA分别对谷氨酰胺、大肠杆菌、肠杆菌和结核分枝杆菌的最终预测没有贡献。总体而言,除AAI外,其余不同物种间的特征贡献差异较大,说明这些物种中的Kace位点可能具有不同的特征。
使用独立测试进行性能验证
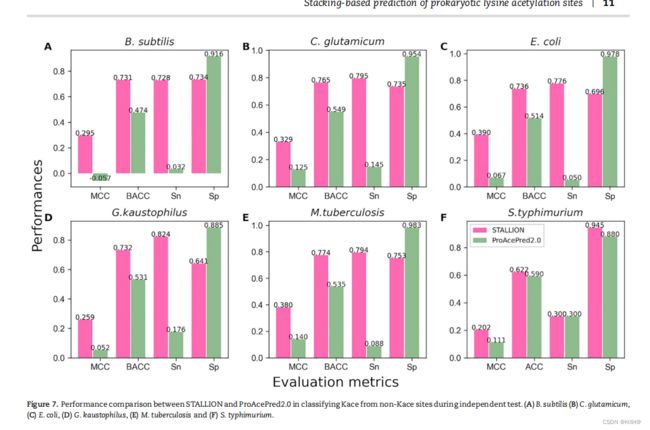
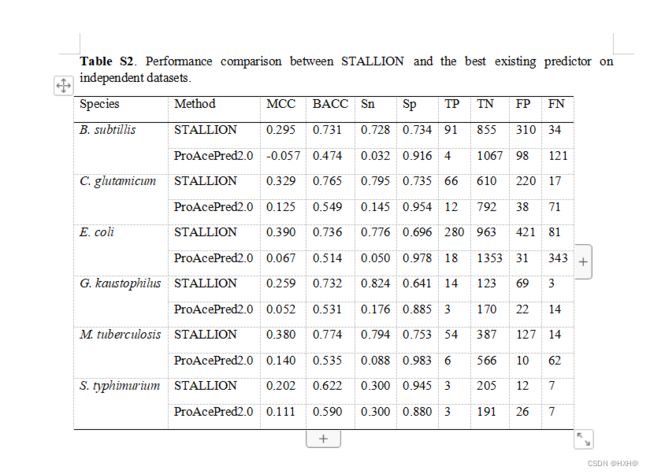
我们进一步使用独立的数据集评估了STALLION,并将其性能与现有的方法进行了比较。自2009年以来,一些计算工具已经被报道用于Kace站点预测。值得注意的是,Chen等人最近使用一个独立的数据集评估了物种特异性ProAcePred 2.0预测器,并与现有方法的性能进行了比较,包括物种特异性ProAcePred,一般预测因子,即。集成桶,PSKAcePred,佛西达和PLMLA。结果显示,ProAcePred 2.0显著优于通用预测因子和他们之前的版本ProAcePred。因此,本研究只考虑ProAcePred 2.0进行比较,排除其他方法,原因如下:(i)物种特异性预测与通用预测比较不公平,这从以往的研究[36,37]可以明显看出,(ii)ProAcePred 2.0是ProAcePred的升级版本。值得一提的是,每个物种的独立数据集都提交给了ProAcePred 2.0 web服务器(http://computbiol.ncu.edu.cn/PAPred),预测是根据给定的默认阈值计算的。值得注意的是,ProAcePred 2.0返回Kace站点及其预测概率值,但不返回非Kace的预测概率值。因此,用部分概率信息计算AUC值可能是不可行的。然而,我们比较了两种方法在MCC方面的性能,这是一个直观和直接的度量,而处理一个不平衡的数据集,如所述。我们的评估结果显示,STALLION的MCC分别为0.295、0.329、0.390、0.259、0.380和0.202 B. 精细的 C. 谷氨酰胺、大肠杆菌、嗜毛孢杆菌、结核分枝杆菌和鼠伤寒杆菌(图7和补充表S2)。STALLION的表现分别优于ProAcePred。5种物种的MCC值为20.0% (B. subtilis, C. glutamicum, E. coli, G. kaustophilus and M. tuberculosis)MCC值为9.1%。STALLION比ProAcePred2.0具有更好的性能:(i)与ProAcePred2.0不同,我们根据系统分析的堆叠框架中排除了KNN特征编码,该分析确定了交叉验证过程中KNN编码的过拟合性质(见下文部分)(2);(ii)与ProAcePred2.0简单的特征选择方法不同,我们采用了一个严格的过程,利用三个不同的评分函数和SFS独立识别每个分类器的最优特征集,这是耗时的,(iii)不像ProAcePred2中的单一模型。我们的堆叠策略集成了5个基于树的集成基线模型,从而导致更准确的Kace站点预测。
就像STALLION和最好的基于单一特征的模型的交叉验证性能比较一样,我们进行了独立的测试。图8显示,所有6种的MCC均优于单一特征模型,其MCC高于2.39–10.68% B. 枯草属植物,1.18–6.08%较高 C. 谷氨酰胺,大肠杆菌高4.0-9.5%,3.51–10.89%高2.5-8.7%,结核分枝杆菌3.51–10.89%高,鼠伤寒杆菌11.29–19.54%高。这些结果再次强调了我们的系统方法在模型构建中的意义。
在Kace预测中,KNN编码的过拟合性质
KNN特征编码被广泛应用于PTM位点的识别,包括以往的Kace位点预测方法。与之前的研究类似,我们也将其纳入了堆叠框架中的11个编码中。初步结果表明,所有物种模型的预测性能均有显著提高在交叉验证期间与 STALLION合作。然而,独立数据集对应的模型性能略优于随机预测,且明显低于 STALLION。因此,我们从堆叠框架( STALLION)中排除了KNN编码。为了更好地理解这一现象,我们为每个物种开发了基于knn的五种树状模型,并进行了检验交叉验证和独立验证性能(表2)。结果表明,5个分类器中有4个(RF、ERT、AB和XGB)的性能相似,略优于GB,平均auc分别为0.895、0.901、0.888、0.888、0.895和0.872 B. 精细的 C. 谷氨酰胺、大肠杆菌、肠杆菌、结核分枝杆菌和鼠伤寒杆菌。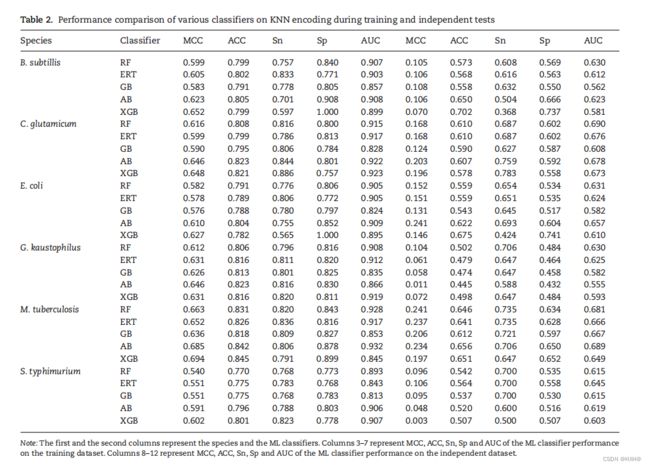
6种植物的独立检验指标分别为0.602、0.665、0.621、0.597、0.670和0.619。6个物种的训练数据集与独立数据集之间的性能差异(AUC差异)从22.46显著增加到29.32%,明显表明无论分类器如何,训练过程中对KNN编码的高估。由于KNN编码方案的过拟合特性,我们强烈建议在将KNN编码方案合并到任何需要大量计算的计算框架中之前,先测试KNN编码的可移植性。
结论
本研究提出了一种堆叠框架,用于准确预测六种不同原核生物的Kace位点。STALLION采用了11种不同的特征编码方案(分为3组)来编码蛋白质片段。随后,采用严格的特征选择方法,为五种不同的基于树的集成算法仔细选择最优特征集,并为每个物种构建各自的基线模型。最后,得到5个基线模型,这些模型经过适当的分类器训练,建立稳定的堆叠STALLION模型。我们提出的方法STALLION在6个不同物种的独立数据集上识别Kace位点方面优于目前最先进的预测器。预计STALLION方法和一个基于堆叠的6个原核生物物种模型的用户友好的web服务器将加快对假定的Kace位点的发现,并极大地帮助更广泛的研究社区的功能表征。我们的研究确定了异质性和互补的特征我们将不断尝试研究其他信息特征,检查它们的贡献,并完善我们的预测平台。总体而言,STALLION方法在Kace站点预测方面取得了稳健的性能,其预测性能需要在几个方面进一步改进。最近报道了新的计算框架,包括基于dl的混合框架[86]和基于dl的方法,自动生成特征。在未来,我们将研究这些方法的可能性,并选择合适的方法来进一步提高Kace站点的预测性能。