R语言limma包差异表达分析
目录
一、数据准备
1.数据加载
2.做分组信息数据
3.表达数据样本ID顺序与样本信息数据匹配
二、数据预处理
(1)缺失值处理
(2)离群值处理
(3)数据归一化
三、数据探索
(1)查看数据是否经过了log2转换
(2)查看管家基因的表达量
(3)画箱线图查看数据分布
(4)PCA图、层次聚类图
四、差异表达分析
(1)数据准备
(2)差异分析及可视化
(3)提取差异表达基因
一、数据准备
1.数据加载
#数据表达数据加载
exp1=read.csv('F:\\bioinformatics\\research program\\materialXXXXXXXXXX\\20221101XXXXXXXXXX\\datafile\\GSE5281\\GSE5281traned.csv')
dim(exp1) #查看数据维度
[1] 20857 162
#加载样本信息数据
sample_info=read.csv('F:\\bioinformatics\\research program\\materialXXXXXXXXXX\\20221101XXXXXXXXXX\\datafile\\GSE5281\\GSE5281 sample.csv')
dim(sample_info) #查看数据维度
[1] 161 122.做分组信息数据
提取样本数据关键信息(分组信息)
sample_data=sample_info[,c(1,2)] #提取样本信息数据的关键数据
head(sample_data) #查看数据
Accession Title
1 GSM238763 EC_affected_1
2 GSM238790 EC_affected_2
3 GSM238791 EC_affected_3
4 GSM238792 EC_affected_4
5 GSM238793 EC_affected_5
6 GSM238794 EC_affected_6
新建修改过的样本分组列
sample_data1=sample_data
sample_data1$group_list=c(rep('AD',87),rep('Control',74))
head(sample_data1) #查看数据
Accession Title group_list
1 GSM238763 EC_affected_1 AD
2 GSM238790 EC_affected_2 AD
3 GSM238791 EC_affected_3 AD
4 GSM238792 EC_affected_4 AD
5 GSM238793 EC_affected_5 AD
6 GSM238794 EC_affected_6 AD删除第二列
sample_info1=sample_data1[,-2] #删除第二列
head(sample_info1) #得到分组信息
Accession group_list
1 GSM238763 AD
2 GSM238790 AD
3 GSM238791 AD
4 GSM238792 AD
5 GSM238793 AD
6 GSM238794 AD查看两个数据长度
length(colnames(exp1)) #表达数据的列数,多了一列gene symbol ID
length(sample_info1[,1]) #样本分组类型的长度
[1] 162
[1] 1613.表达数据样本ID顺序与样本信息数据匹配
表达数据中的列名样本ID号是长成这样的:
colnames(exp1)[2] #随便看一一个
[1] "X.GSM119615."为了匹配,需要先修改一下
list1<-list() #新建一个空列表
cols=colnames(exp1[,2:ncol(exp1)]) #提取表达数据的所有样本ID
cols[2] #确认字符串样式
[1] "X.GSM119616."循环语句进行修改
for (i in c(1:length(cols))) {
list1[i]=substr(cols[i],3,11)
}
head(list1) #查看数据
[[1]]
[1] "GSM119615"
[[2]]
[1] "GSM119616"
[[3]]
[1] "GSM119617"
[[4]]
[1] "GSM119618"
[[5]]
[1] "GSM119619"
[[6]]
[1] "GSM119620"替换修改后的样本ID
exp2=exp1
names(exp2)[2:ncol(exp1)]<-list1 #修改列名查看数据
head(colnames(exp2))
head(sample_info1[,1])
[1] "symbol" "GSM119615" "GSM119616" "GSM119617" "GSM119618"
[6] "GSM119619"
[1] "GSM238763" "GSM238790" "GSM238791" "GSM238792" "GSM238793"
[6] "GSM238794"确认两个集合相等
#样本ID比对
FT=colnames(exp2[,2:ncol(exp2)]) %in% list1
a=0
for (j in FT) {
if (j==TRUE) {
a=a+0
}
else if (j==FALSE) {
a=a+1
}
}
a #a=0说明样本对应没有问题解决顺序的问题
###修改样本对应顺序,让两个数据的样本数据一致
sample_info2=sample_info1[match(list1,sample_info1$Accession),]
head(sample_info2 ) #现在两个数据的样本信息数据就一致啦
Accession group_list
88 GSM119615 Control
89 GSM119616 Control
90 GSM119617 Control
91 GSM119618 Control
92 GSM119619 Control
93 GSM119620 Control二、数据预处理
(1)缺失值处理
dim(exp2)
sum(is.na(exp2)) #没有数据缺失
##若有数据缺失,直接过滤就行
#new_data=na.omit(exp2)(2)离群值处理
#数据离群处理
#处理极端值
#定义向量极端值处理函数
dljdz=function(x) {
DOWNB=quantile(x,0.25)-1.5*(quantile(x,0.75)-quantile(x,0.25))
UPB=quantile(x,0.75)+1.5*(quantile(x,0.75)-quantile(x,0.25))
x[which(xUPB)]=quantile(x,0.5)
return(x)
}
head(names(exp2))
#第一列设置为行名
exp3=exp2
rownames(exp3)=exp2[,1]
exp4=exp3[,-1]
head(exp4)
#处理离群值
exp5=apply(exp4,2,dljdz) (3)数据归一化
#数据归一化
library(limma)
exp6=normalizeBetweenArrays(exp5)
三、数据探索
(1)查看数据是否经过了log2转换
head(exp6) #发现数据并未经过对数转换发现数据很大,并未经过log2对数转换
exp7=data.frame(exp6)
#做对数转换
exp8=mutate_if(exp7,is.numeric,funs(log2)) #对数据做对数转换
head(exp8)
head(exp7)
[1] 20857 161
[1] 20857 161(2)查看管家基因的表达量
#1管家基因的表达量
exp8['GAPDH',] #挺高的:GAPDH
exp8['ACTB',] #也挺高的:ACTB
(3)画箱线图查看数据分布
画图数据准备
################画出各种图看看
library(reshape2)
exp8_L=melt(exp8) #宽数据转为长数据
head(exp8_L)
dim(exp8_L)
names(exp8_L)[1]='sampleID' #修改列名
head(exp8_L) #查看数据
####将分组信息加入长数据
library(stringr)
str_detect(sample_info2$group_list,'Control')
g_list=ifelse(str_detect(sample_info2$group_list,'Control')==TRUE,'Control','AD')
length(g_list)
head(g_list) #看看g_list是个什么东西?
exp8_L$group=rep(g_list,each=nrow(exp8))
head(exp8_L) #查看数据
dim(exp8_L) #查看数据维度
##加入分组信息到长数据中完毕画箱线图查看数据分布情况
###接下用ggplot2画图
library(ggplot2)
p=ggplot(exp8_L,
aes(x=sampleID,y=value,fill=group))+geom_boxplot() #fill参数:用分组进行颜色映射
print(p) GSE5281箱线图
GSE5281箱线图
上面的图还可以进一步精修
##箱线图精修版
p00=ggplot(exp8_L,
aes(x=sampleID,y=value,fill=group))+geom_boxplot() #fill参数:用分组进行颜色映射
#去除网格线和背景
p00=p00+theme_bw()+theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
axis.line = element_line(colour = 'black'))
p00 #查看图形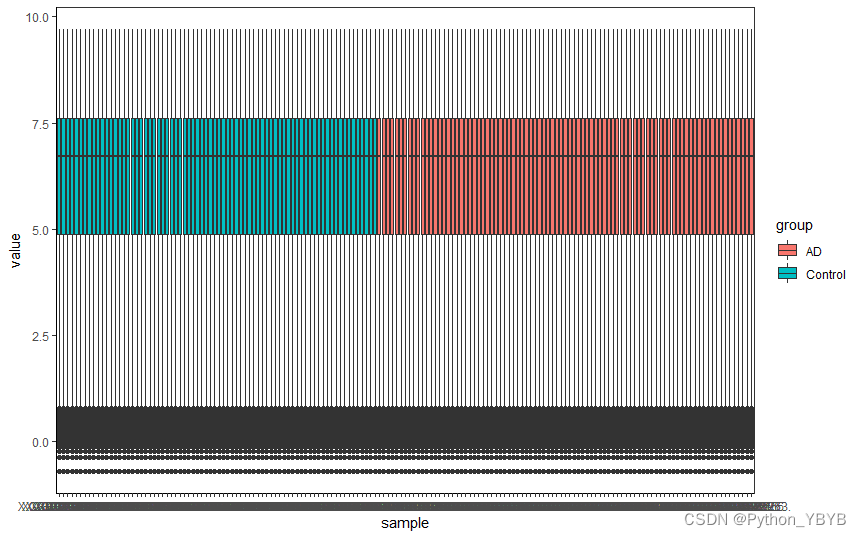 GSE5281箱线图(精修版)
GSE5281箱线图(精修版)
(4)PCA图、层次聚类图
层次聚类图
#层次聚类图
sample_info2
exp9=exp8
colnames(exp9)=paste(sample_info2$group_list,1:ncol(exp8),sep = '')
head(exp9)
#定义nodePar
nodePar=list(lab.cex=0.6,pch=c(NA,19),cex=0.7,col='blue')
#聚类
hc=hclust(dist(t(exp9))) #t()的意思是转置
#绘图
plot(as.dendrogram(hc),nodePar = nodePar,horiz = TRUE) GSE5281层次聚类图
GSE5281层次聚类图
好像看不出来什么。。。。。。。。。。。。。。。。。。。。
PCA图
##PCA图
library(ggfortify)
df=as.data.frame(t(exp8)) #转置后就变成了矩阵
dim(df) #查看数据维度
dim(exp8)
df$groupp=sample_info2$group_list #加入样本分组信息
autoplot(prcomp(df[,1:ncol(df)-1]),data=df,colour='groupp') #PCA散点图
 GSE5281 PCA散点图
GSE5281 PCA散点图
大致是分开了
四、差异表达分析
(1)数据准备
需要三个数据:表达矩阵已经有啦(exp6);还有:分组矩阵,差异比较矩阵
分组矩阵
design=model.matrix(~0+factor(sample_info2$group_list))
colnames(design)=levels(factor(sample_info2$group_list))
head(exp6)
row.names(design)=colnames(exp6)
design #得到分组矩阵:0代表不是,1代表是
AD Control
GSM119615 0 1
GSM119616 0 1
GSM119617 0 1
GSM119618 0 1
GSM119619 0 1
GSM119620 0 1差异比较矩阵
##差异比较矩阵
contrast_matrix=makeContrasts(paste0(c('AD','Control'),collapse = '-'),levels = design)
contrast_matrix #-1和1的意思是Control是用来被比的,AD是用来比的
Contrasts
Levels AD-Control
AD 1
Control -1
> (2)差异分析及可视化
分三步走:lmFit;eBayes;topTable
step1
#step:lmFit
fit=lmFit(exp8,design)
fit2=contrasts.fit(fit,contrast_matrix)step2
#step:eBayes
fit3=eBayes(fit2)step3
step3:topTable
tempoutput=topTable(fit3,coef = 1,n=Inf)
DEG_M=na.omit(tempoutput) #得到差异分析矩阵,重点看logFC和P值
head(DEG_M) #查看数据
logFC AveExpr t P.Value adj.P.Val B
NEAT1 3.872485 9.512774 15.17968 4.053906e-33 8.455232e-29 64.36975
MSI2 1.621265 9.321856 12.00464 2.962532e-24 2.779383e-20 44.43212
EPC1 1.476923 9.952469 11.91114 5.412282e-24 2.779383e-20 43.84258
DTNA 1.873689 12.034226 11.90096 5.779352e-24 2.779383e-20 43.77838
AMER2 1.406314 12.314261 11.85761 7.641583e-24 2.779383e-20 43.50512
NAV2 1.329965 9.248507 11.85058 7.995541e-24 2.779383e-20 43.46082接下来就是将上述结果可视化
热图:选取前40个最显著的基因做热图,查看差异是否真的很显著
library(pheatmap)
f40_gene=head(rownames(DEG_M),40)
f40_subset_matrix=exp6[f40_gene,]
head(f40_subset_matrix)
f40_subset_matrixx=t(scale(t(f40_subset_matrix))) #数据标准化。。。数据标准化和归一化的区别:平移和压缩
pheatmap(f40_subset_matrixx) #出图 GSE5281前40个差异基因表达数据热图
GSE5281前40个差异基因表达数据热图
火山图
火山图1,没有显示差异基因
#火山图
colnames(DEG_M)
plot(DEG_M$logFC,-log10(DEG_M$adj.P.Val)) #查看图形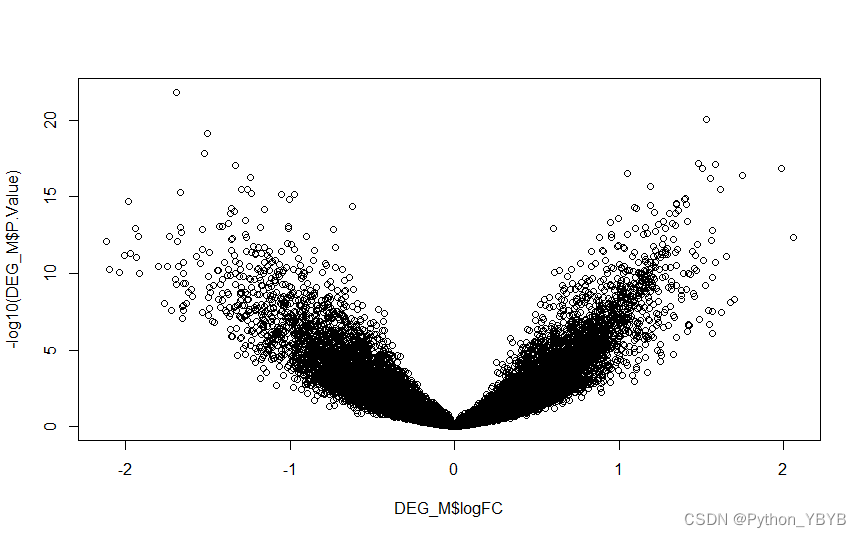 GSE5281火山图
GSE5281火山图
火山图2,显示差异基因
DEG=DEG_M
logFC_cutoff=0.5 #选差异倍数的阈值
logFC_cutoff
####给差异分析矩阵数据中的基因加上标签
DEG$change=as.factor(ifelse(DEG$adj.P.Val<0.05 & abs(DEG$logFC)>logFC_cutoff,
ifelse(DEG$logFC>logFC_cutoff,'UP','DOWN'),'NOT'))
head(DEG) #查看数据
nrow(DEG[DEG$change=='UP',]) #上调基因数量
nrow(DEG[DEG$change=='DOWN',]) #下调基因的数量
dim(DEG)
dim(exp6)
thttile=paste0('GSE',5281,
'\nCutoff for logFC is ',round(logFC_cutoff,1),
'\nThe number of up gene is ',nrow(DEG[DEG$change=='UP',]),
'\nThe number of down gene is ',nrow(DEG[DEG$change=='DOWN',]))
thttile #查看准备的火山图标题
g=ggplot(data = DEG,aes(x=logFC,y=-log10(adj.P.Val),color=change))+
geom_point(alpha=0.4,size=1.75)+
theme_set(theme_set(theme_bw(base_size = 20)))+
xlab('log2 fold change')+ylab('-log10 adj.P.Val')+
theme_bw()+theme(panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_blank(),
axis.line = element_line(colour = 'black'))+
ggtitle(thttile)+theme(plot.title = element_text(size = 15,hjust = 0.5))+
scale_color_manual(values = c('blue','black','red'))
print(g) #输出火山图 GSE5281差异分析火山图
GSE5281差异分析火山图
(3)提取差异表达基因
head(DEG)
rownames(DEG[DEG$change=='UP' | DEG$change=='DOWN',])
DEG_MATRIX=exp8[row.names(exp8) %in% rownames(DEG[DEG$change=='UP' | DEG$change=='DOWN',]),]
dim(DEG_MATRIX) #差异表达基因表达数据
UP_DEG=exp8[row.names(exp8) %in% rownames(DEG[DEG$change=='UP',]),]
dim(UP_DEG) #上调基因表达数据
DOWN_DEG=exp8[row.names(exp8) %in% rownames(DEG[DEG$change=='DOWN',]),]
dim(DOWN_DEG) #下调基因表达数据
write.csv(DEG_MATRIX,
'F:\\bioinformatics\\research program\\materialXXXXXXXXXX\\20221101XXXXXXXXXX\\datafile\\GSE5281\\DEG_MATRIXGSE5281.csv')
write.csv(UP_DEG,
'F:\\bioinformatics\\research program\\materialXXXXXXXXXX\\20221101XXXXXXXXXX\\datafile\\GSE5281\\UP_DEGGSE5281.csv')
write.csv(DOWN_DEG,
'F:\\bioinformatics\\research program\\materialXXXXXXXXXX\\20221101XXXXXXXXXX\\datafile\\GSE5281\\DOWN_DEGGSE5281.csv')
好啦,就到这里,欢迎点进来的同学交流和学习!