r语言删除csv中na行_R 利用 limma 包作简单的差异基因分析
下载、整理 GEO 预处理 Matrix 文件
首先从 GEO 站点 下载 GEO matrixfile 数据压缩包,并将其解压到 R 的当前工作目录。
获取表达值矩阵文件
打开类似 GSEXXXX_series_matrix.txt,然后将文件中每行首位中含有 ! 的行全部删除,只留下从 ID_REF 开始的数据行,然后将其另存为 GSEXXXX_matirx.txt 文件。后续通过 Excel 将其保存为 .csv 格式文件。
读入获取的表达值矩阵文件以及样本分组文件
BiocManager::install('limma')library(limma)# 读入获取的表达值矩阵文件expr= read.csv("GSEXXXX_matrix.csv", header= TRUE, row.names= 1)str(expr)# 读入样本分组文件pacman::p_load(xlsx)sample= read.xlsx('SAM.xlsx', sheetName = 'Sheet1', header = FALSE)nrow(sample) #63# 根据样本分组文件信息筛选特定的63个样本gene_expr= subset(expr,select = colnames(expr) %in% sample[,1])str(gene_expr)summary(gene_expr) #观察数值范围判断是否需要对表达值进行对数转换
qx= as.numeric(quantile(gene_expr, c(0., 0.25, 0.5, 0.75, 0.99, 1.0), na.rm= TRUE))LogC= (qx[5] > 100) | (qx[6] - qx[1] > 50 & qx[2] > 0) | (qx[2] > 0 & qx[2] < 1 & qx[4] > 1 & qx[4] < 2)if(LogC){ gene_expr[which(gene_expr <= 0)]= NaN gene_exprSet= log2(gene_expr) print("log2 transform needed")}else{ print("log2 transform not needed")}sum(gene_expr < 0) #判断表达值中是否存在负值观察样本间的基因表达数据一致性
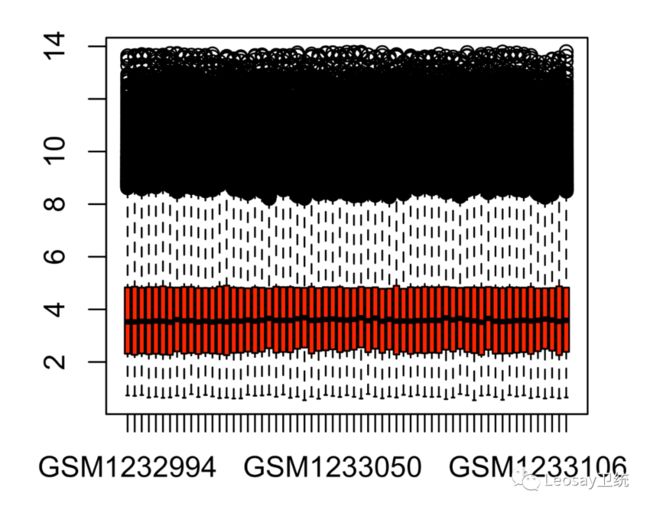
boxplot(gene_expr, col= "red") #基于箱线图进行数据整体情况观察样本间的基因表达数据一致性# 若基因表达数据一致性不是太好,可采用limma包中normalizeBetweenArrays函数来标准化数据norm_gene_expr= normalizeBetweenArrays(gene_expr)group= sample[,2] #获取第二列的分组信息table(group) #pCR: 31, RD: 32design= model.matrix(~ 0 + factor(group)) #将分组进行0和1因子转换colnames(design)= levels(factor(group)) #将分组作为列rownames(design)= colnames(norm_gene_expr) #将表达值作为行# 观察样本分组是否一一对应:head(design)head(sample)基因差异分析
matrix= makeContrasts(paste0(unique(group), collapse= "-"), levels= design) #makeContrasts构建比较矩阵matrixfit= lmFit(norm_gene_expr, design) #为每个基因进行线性模型拟合fit2= contrasts.fit(fit, matrix) #基于拟合的线性模型进行系数估计和标准误计算fit3= eBayes(fit2) #基于经验贝叶斯进行t统计量、F统计量和差异对数表达计算output= topTable(fit3, coef=1, n=Inf, adjust="BH") #对线性模型拟合结果进行排序,提取排序靠前的基因列表DEGs = na.omit(output) #删除基因排序列表中出现的缺失值write.table(DEGs, "DEGs.csv", quote=F, sep=",") #得到差异筛选结果文件火山图可视化
给定logfc绝对值 ≥ 0.3、p < 0.05为有差异的标准。
DEGs$threshold[DEGs$P.Value < 0.05 & DEGs$logFC >= 0.3]= "up"DEGs$threshold[DEGs$P.Value < 0.05 & DEGs$logFC <= -0.3]= "down"DEGs$threshold[DEGs$P.Value >= 0.05 | (DEGs$logFC > -0.3 & DEGs$logFC < 0.3)]= "non"ggplot(DEGs,aes(x= logFC,y= -log10(P.Value),colour= threshold)) + xlab("log2 Fold Change") + ylab("-log10 P.Value") + geom_point() + scale_color_manual(values =c("#6495ED","red","#FF4500")) + geom_hline(yintercept=1.30103,linetype=3) + geom_vline(xintercept=c(-0.5,0.5),linetype=3) + theme(legend.title = element_blank(),panel.grid.major = element_blank(),panel.grid.minor = element_blank())
精彩笔记系列
统计学习笔记 R 语言学习笔记 SAS 学习笔记 Linux 学习笔记 Python 学习笔记 线代学习笔记