木桶排序算法_【生信常识】二代测序的比对算法浅析
前言
本来我只打算将孟大哥的视频内容做一个文字版的概述,然后孟大哥说,不如再加一个算法推导吧,然后我就开始看多一些东西,然后就想着把孟大哥视频里面大概提及然后没有仔细讲的部分做一些补充,完善整个体系的浅层内容学习,所以就有了本文。
废话不多说,我们开始本次的主题吧~
相信大家日常跑数据的时候总是要从检查数据质量,比对,转换格式,差异分析这么一套既定的流程对我们的rna-seq或者chip-seq数据进行处理,然后写报告,发paper(醒醒~,你实验还没补完呢)。
而在这些步骤里面,将原始数据比对到参考基因组上这一步显得尤为重要,这一步的好坏对后面的影响较大(你咋不suo测序质量的好坏影响更大呢~好吧,越上游的东西其实对后面的影响会越大),故而了解比对算法的原理,然后根据自己数据的类型选择合理的工具,显得尤为重要(你这语气是在写申请书吗。。。。。)
现在主流的比对软件不下十种,但按照核心算法区分,其实可以拆分成为两大阵营:
1.基于哈希表(hash-table)数据结构的比对算法
2.Burrows Wheeler transform(BWT)索引数据结构的比对算法
基于哈希表数据结构的比对算法
hash-table的核心思想就是采用种子序列定位及延伸算法(seed-and-extend algorithm)
根据索引构建对象的不同,可以分为两类,第一种,基于参考基因组(reference genome)索引的的延伸比对
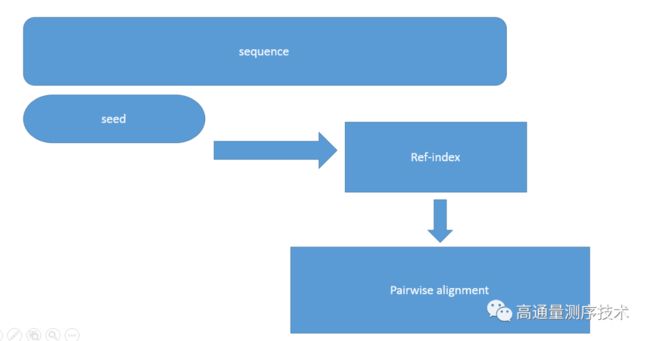
通过检索短序列数据集的hash-table,然后找到ref跟reads完全匹配(match)的子序列,即seed位点,而后通过经典的全局比对或者局部比对,将整条reads比对上去
代表软件是SOAP,PASS,GASST
第二种,基于短序列数据集索引的延伸比对
这回就反过来了,对整个短序列进行索引的构建,然后通过查询ref的hash数据结构,从而得到短序列在ref上的匹配候选点
代表软件:MAQ,SeqMap
而根据比对策略不同,又可以分为两大类,连续种子序列(contiguous seed)跟间隔种子序列(spaced seed)
首先,我们来说一下连续种子序列策略

先将短序列分为k-mer长的重叠子序列
然后查询由基因组k-mer长的子序列构成的hash-table进行匹配
如果完全match上,则短序列定位到ref的该位点
这种方法的缺点是显而易见的,即不能允许mismatch的存在,如果序列中出现至少一个位点的突变,则该位点就会被过滤掉
而为了弥补这种缺陷,设计者们找到了一个解决的方法:鸽洞原理(pigeonhole principle)
具体方法如下:
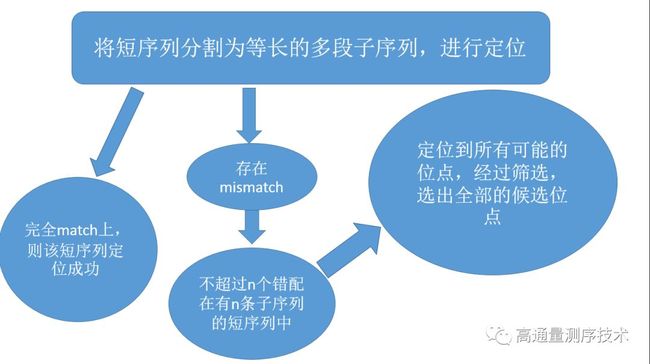
由此增加了对mismatch的容错率
连续种子序列的缺点就在于随着过多的mismatch的出现,需要将序列分割成更短的子序列,而过短的子序列会mapping到参考基因组上的很多位置,从而造成效率低下,故而出现了所谓间隔种子序列(spaced seed)策略
间隔种子序列,说大白话就是,种子序列中间存在若干个不确定碱基,即在查找过程中允许mismatch的存在,举个栗子,间隔种子序列AGxCGTAA,既可以跟AGGCGTAA匹配,也可以跟AGCCGTAA匹配。这样做的后果就是增加了比对计算时间复杂度,但优点在于增强了比对算法的灵敏度。
而对于上面两种比对策略,其实每个软件都有不同的侧重点,有些拥有两种的混合模式进行匹配,从而在时间跟准确度上取得一个平衡。
Burrows Wheeler Transform(BWT)索引数据结构的比对算法
讲完基于hash-table的比对算法,大家应该能够能深刻感受到,如果我们处理的数据里面存在大量重复序列的话,那对于这一类软件来说简直就是噩梦,因为我们并不能知道这些重复序列的相对位置,故而后面在设计软件的人开始了新的思路,即采用了后缀树(suffix tree)和后缀矩阵(suffix array)索引从而解决了重复序列的比对问题。但于此带来的是内存消耗量的显著提升,故而这类算法的软件一般只用于处理基因组较小的生物数据。
而后,采用了BWT算法的软件应运而生,现在让我们来了解一些什么是BWT算法还有它在数据处理中是如何应用的BWT (Burrows–Wheeler_transform)数据转换算法:
将原来的文本转换为一个相似的文本,转换后使得相同的字符位置连续或者相邻,之后可以使用其他技术如:Move-to-front transform 和 游程编码 进行文本压缩。
该算法一开始是用于数据压缩的,而后应用于生物学比对中,而具体实现步骤如下:


这样就完成了一个转置矩阵,如果进行压缩的话,像上面转置后矩阵的最后一列三个A,可以记做Ax3,在实际压缩中如果存在很多相同的字符,这样压缩很明显节约了很多空间。
接着进行解码还原的操作:
1.L列的第一个元素为原始序列的最后一个元素(因为$在该位置后面)。
2.F列中的每一个元素,都是其同一行中的L列的下一个元素。也就是L列是F列的前一个元素。
看着有些晕,不如看下面是怎么搞的吧



而当实际应用时,可能你拿去匹配的是该序列的其中一小部分,那么bwt算法又是怎么样来避免错配的呢,我们可以看下面的栗子:


如果看不太明白,可以自己手动推演一下,再不明白,可以去看看孟大哥的板演过程.
而类似hash-table的连续种子序列策略,这种寻找seed的方法并不能容忍mismatch的存在,更不用说gap之类的存在,而为了解决这一方面的问题,软件设计者们通过两种方法进行避免,从而提高了灵敏度,这两种方法如下:
1) 假设一段序列是20bp,那么将其拆解为18,2两个片段,也就是说这个前18bp如果完全match上了,后面的2bp即使match不上,我们也认为它是候选seed,至于这个window的大小,是根据自己的条件选择,这样做的缺点仍然是在高变区域seed难以找到。

2)假设一段子序列为16bp,如果后面的10bp跟下一段子序列的前10bp重叠,那么就认为是候选的seed,这样就从另一个方面提高了容错率。
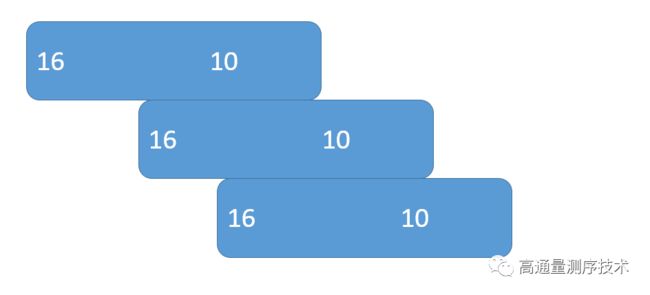
(这里插入一下,感谢重蓬飞老哥的指正,重老哥说这里应该提及一下LF-mapping的原理,并且给出了资料,请各位看一下这个文档)
https://www.cs.jhu.edu/~langmea/resources/lecture_notes/bwt_and_fm_index.pdf
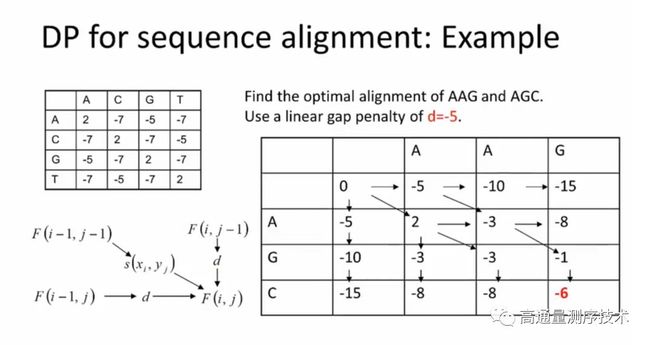
这样就完成了两大比对算法的原理讲解,在通过seed找到定位之后,就是通过局部比对跟全局比对的方法对整条序列进行匹配,而匹配的原理,这里放高歌老师在公开课上的ppt截图,有兴趣了解的可以去看一下北大那个生信公开课对这两个经典比对算法的讲解


之前,孟大哥跟我说,既然要写这个东西,不如加个推导会比较好玩一些,然后我就去了解了一些相关的东西,然后在正月点灯笼的视频里面,看到一个有趣的东西:
Suffix array = 1- BWT
也就是说在相同时间里,如果创建了一个suffix array,那同时也相当于创建了一个bwt,所以我就跑去了解suffix array是什么东西,在这里,跟大家分享一下两个关于后缀数组(suffix array)的博客(我尝试写了一些,发现还是别人讲得比较透彻,所以自暴自弃~)
那么如何理解后缀数组呢,首先我们先来了解一些,什么是基数排序

看起来感觉很蒙对吧,那么看接下来是实际上操作是怎么一回事吧
首先,我们给定一些数值,73, 22, 93, 43, 55, 14, 28, 65, 39, 81
接着假定我们有十个木桶,序号从0到9,我们将个位数相同的数字摆进相同的桶里面

接着,将这些数字按照上面得到的顺序重新排列,得到
81, 22, 73, 93, 43, 14, 55, 65, 28, 39
然后,我们现在又有十个桶,将这些数字以十位数为排序标准,依次放进这些桶里面
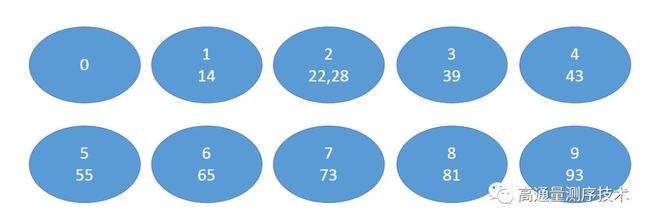
再将上面得到的顺序排列下了,得到新的数组
14, 22, 28, 39, 43, 55, 65, 73, 81, 93
这样就完成了依次基数排序,从而我们就得到了这个原始数组里面各位值的大小关系,其实后缀数组也是完成类似的东西,通过排序得到数组内各数值的相关关系
接着,我们来正式了解一下后缀数组
首先,我们必须了解的关于后缀数组的两大关键部件
1)SA[i],这个什么东西呢?这东西就叫后缀数组,它是一个一维数组,保存1..n 的某个排列 SA[1] ,SA[2] , ……, SA[n] ,将 S 的 n 个后缀从小到大进行排序之后把排好序的后缀的开头位置顺次放入SA 中。
2)rank[i],以下标i开头的后缀在所有后缀中从小到大排列的 “ 名次 ”,决定着每个元素应该在什么位置出现。
怎么去简单理解这两个东西,rank[i]决定排第几,SA[i]决定第几的元素是谁
如果我们去查百度百科,我们会得到这样一张图

怎么样去理解这一张图呢,请跟着我的思路过来
首先,给定一个字符串,以上面的为例

接着,我们给每个字符减去(a-1),即得到这样的一个结果

接着,我们将所有的后缀数组写出来

看到这里熟不熟,其实在后面加个$并且补全,不就是上面的BWT矩阵吗?
好的,我们接着说下去
得到

之后,我们将每一个数与它的下一位排名组合成一个新的组合,即得到在时间复杂度O(nlogn)当n为0时的数组

将得到的这个新的数组按基数排序进行排序,最后得到他们的排名的情况如下:
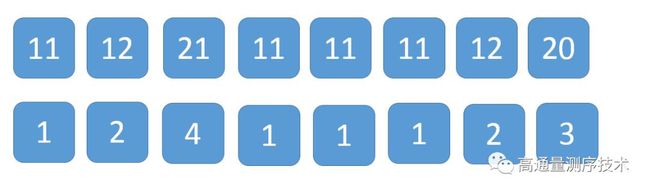
这样其实我们就已经大概知道了,a后面要么是a(11),要么是b(12),而b后面要么是a(21),要么没有(20),但我们还不知道a与a之间的相对位置等信息,所以接下来,我们将得到的新数组的排名以两个数字为移动单位,进行串联,形成新的数组,即

原本1,其实相当于原本11(aa)的简化,同理知道2为12(ab),4为21(ba),3为20(b),而这样以两个数字为锚定单位,其实就是进一步确定信息,即1(aa)和4(ba)谁前谁后,同理知道其他各组合的相对位置,这里你们可以自己看看,有哪些位置信息已经被确认,毕竟这不能纯粹变成一篇爽文,需要加以思考才行
然后接着对新的数组进行基数排序,我们得到了新的一个排名

通过这样,其实每个部分的相对位置其实就已经确认了,谁先谁后已然知晓
本文的参考材料如下
[1]苏州大学硕士尚婧的毕业论文《下一代测序短序列比对软件算法比较及评价》
[2]坑主孟浩巍b站视频:20171026-基于BWT算法的比对软件原理解析(BWA & Bowtie & Bowtie2)
[3]b站up正月点灯笼视频:Suffix array简介和构建
[4]博客园博主I'MJACKY博客
以上为本人学习相关内容的资料,有兴趣仔细研究的小伙伴可以自行到相关网站上面搜索与学习,他们都研究的比较熟路,我只是本着大概了解的态度对上面这些大佬的内容进行很粗浅的学习,并且汇集成本文内容,然后跟大家大概介绍一下二代测序分析中常用的比对算法大概是怎么作用的~
好了,本期到此结束,希望对您的生信学习有所帮助~