ATAC-seq分析:数据处理(5)
1. 子集划分
我们可能希望将比对的读数分成代表核小体游离和核小体占据的读数。在这里,我们通过使用插入大小来过滤读取,为代表无核小体、单核小体和双核小体的读取创建 BAM 文件。
atacReads_NucFree <- atacReads[insertSizes < 100, ]
atacReads_MonoNuc <- atacReads[insertSizes > 180 & insertSizes < 240, ]
atacReads_diNuc <- atacReads[insertSizes > 315 & insertSizes < 437, ]
2. BAM创建
读取的结果可以写回 BAM 文件,用于我们分析的其他部分,或者通过 rtracklayer 包中的函数在 IGV 等程序中进行可视化。
nucFreeRegionBam <- gsub("\\.bam", "_nucFreeRegions\\.bam", sortedBAM)
monoNucBam <- gsub("\\.bam", "_monoNuc\\.bam", sortedBAM)
diNucBam <- gsub("\\.bam", "_diNuc\\.bam", sortedBAM)
library(rtracklayer)
export(atacReads_NucFree, nucFreeRegionBam, format = "bam")
export(atacReads_MonoNuc, monoNucBam, format = "bam")
export(atacReads_diNuc, diNucBam, format = "bam")
3. 创建 GRanges 片段
我们可以从单端读取中重新创建全长片段,以评估重复率并创建片段的 bigwig。在这里,我们使用 granges() 函数从配对的单端读取中重新创建完整片段。
atacReads[1, ]
 atacReads
atacReads
atacFragments <- granges(atacReads)
atacFragments[1, ]
 atacFragments
atacFragments
我们可以使用 duplicated() 函数来识别我们的全长片段的非冗余(非重复)部分。
duplicatedFragments <- sum(duplicated(atacFragments))
totalFragments <- length(atacFragments)
duplicateRate <- duplicatedFragments/totalFragments
nonRedundantFraction <- 1 - duplicateRate
nonRedundantFraction
4. 创建 bigWig
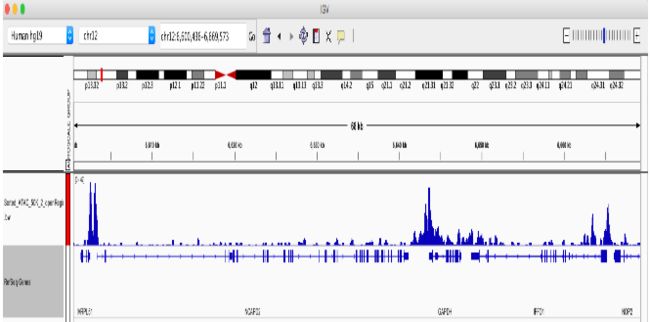
通过创建一个 bigWig 文件,我们可以大大加快在基因组浏览器中查看 ATACseq 信号堆积的速度。此时可以对总映射读取进行额外的标准化。
openRegionRPMBigWig <- gsub("\\.bam", "_openRegionRPM\\.bw", sortedBAM)
myCoverage <- coverage(atacFragments, weight = (10^6/length(atacFragments)))
export.bw(myCoverage, openRegionRPMBigWig)
欢迎Star -> 学习目录
更多教程 -> 转录组测序分析教程合集
更多教程 -> 单细胞系列教程:合集
本文由 mdnice 多平台发布