DLKcat开发细则(自用)
数据预处理
DLKcat模型开发相关内容从代码层面来看有如下几个代码较为重要,

preprocess文件是预处理部分,这部分内容是将数据集中的Smlies和Sequence进行编码并存入一个npy文件中,后在模型的训练(或者预测)阶段,读取这部分保存的内容。具体的运行流程如下所示。
数据集的内容
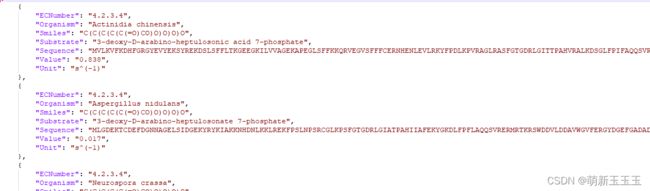

这里我们可以看出数据集所包含的内容,所包含的是1W7千多个的字典,可以通过读取字典的方式去取出其中的内容。
![]()

读取Kcat的文件,smiles就是数据中每个字典所包含的Smiles,sequence就是数据中每个字典包含的Sequence,Kcat是数据中每个字典所含的Value值,这样我们便得到了所需要处理的数据。
原子编码
第一步是将mol模型通过rdkit的包解析出来,这里添加了H元素,因为数据集中smiles都是不考虑手性且不含H元素的。
mol = Chem.AddHs(Chem.MolFromSmiles(smiles))
#打印mol
# mol = Chem.MolFromSmiles(smiles)
# Draw.ShowMol(mol, size=(500, 500), kekulize=False)

通过打印出来的小分子图我们可以比较直观的看出,这个小分子含有,6个O元素,12个H元素和6个C元素。其中,每个与每个原子相连接的部分称为化学键。后续有提取元素并编码,和提取化学键并编码的代码,因此在此做一些说明。
原子编码的代码:
atoms = create_atoms(mol)
create_atoms是自己定义的模块,如下所示:
def create_atoms(mol):
"""Create a list of atom (e.g., hydrogen and oxygen) IDs
considering the aromaticity."""
# atom_dict = defaultdict(lambda: len(atom_dict))
atoms = [a.GetSymbol() for a in mol.GetAtoms()]#获得原子
# print(atoms)
for a in mol.GetAromaticAtoms():
i = a.GetIdx()
atoms[i] = (atoms[i], 'aromatic')
atoms = [atom_dict[a] for a in atoms] #对原子编码
return np.array(atoms)
可见我们首先将mol输入进来后通过GetAtoms这个方法,逐一提取小分子的原子。经过我们之前在图中的计算,一共24个原子,那么通过循环读取可知

mol.GetAromaticAtoms():获取芳香环中的原子
最终通过
atoms = [atom_dict[a] for a in atoms]
对原子进行编码。这里要说明一下,
atom_dict = defaultdict(lambda: len(atom_dict))
这里的defaultdict的用法可以参考
https://blog.csdn.net/jiaxinhong/article/details/108398099
通过编码得到的结果如图所示:
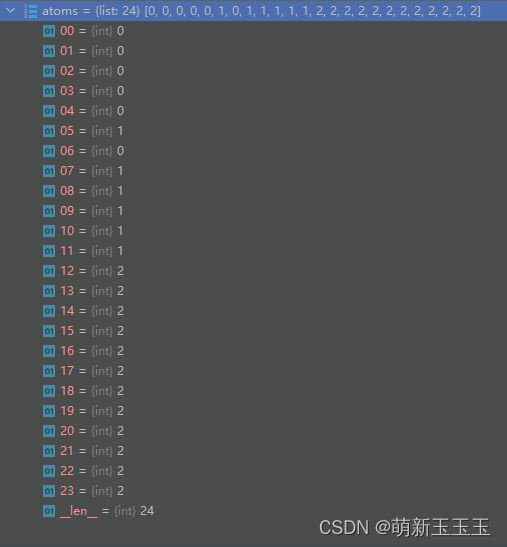
由此可以看出,C元素全部被编码为0,O元素全部被编码为1,H元素全部被编码为2。到此对原子的初步编码就已经完成了。
获取化学键
获取化学键这部分的代码如下所示。什么是化学键在之前已经有所介绍。接下来将详细分析这部分代码所运行的功能。
def create_ijbonddict(mol):
# bond_dict = defaultdict(lambda: len(bond_dict))
i_jbond_dict = defaultdict(lambda: [])
for b in mol.GetBonds():#获取化学键
#bond.GetBeginAtomIdx(),GetEndAtomIdx:获取键两端的原子索引
i, j = b.GetBeginAtomIdx(), b.GetEndAtomIdx()
#GetBondType获取化学键的类型
bond = bond_dict[str(b.GetBondType())]
i_jbond_dict[i].append((j, bond))
i_jbond_dict[j].append((i, bond))
return i_jbond_dict
首先循环mol的化学键,通过GetBonds的方式。然后通过bond.GetBeginAtomIdx(),GetEndAtomIdx:获取键两端的原子索引,再通过字典的形式来获得共价键的字典。此时的共价键分类是单双键的分法,
由这个图可知,此时只有一个双键其余的都是单键。因此最终可得结果为:

这个字典将每个原子与化学键之间的位置表示了出来。
指纹
DLKcat对分子结构进行了一个“指纹”的计算方式,其具体的算法参考了Weisfeiler-Leman算法。简单的来说就是一个不断迭代相邻结点的一个算法值,这里的半径radius值就是迭代的次数。具体的算法操作流程可以参考:
https://blog.51cto.com/u_15080014/2619583
def extract_fingerprints(atoms, i_jbond_dict, radius):
# fingerprint_dict = defaultdict(lambda: len(fingerprint_dict))
# edge_dict = defaultdict(lambda: len(edge_dict))
if (len(atoms) == 1) or (radius == 0):
fingerprints = [fingerprint_dict[a] for a in atoms]
else:
nodes = atoms
i_jedge_dict = i_jbond_dict
for _ in range(radius):
fingerprints = []
for i, j_edge in i_jedge_dict.items():
neighbors = [(nodes[j], edge) for j, edge in j_edge]
fingerprint = (nodes[i], tuple(sorted(neighbors)))
fingerprints.append(fingerprint_dict[fingerprint])
nodes = fingerprints
这里我们可以看到else的部分,我们以一个i_jedge_dict为例。
![]()
由于i_jedge_dict为一个字典,则i为索引数,j_edge为字典所包含的内容,这里可以看出为第一个C原子的化学键。
neighbors = [(nodes[j], edge) for j, edge in j_edge]
neighbors解释:j和edge分别取j_edge中每个元素的两个值,比如这里,j就是1,edge就是0。nodes[1]就是之前元素编码后的nodes的一号元素。
![]()
这里我们可以看出最后的结果。(nodes[11]=1,nodes[12]=2,node[13]=3)最终输出结果为:

我们可以看到通过计算“指纹”的方式,原子本来的编码值发生了变化。这个循环还要进行一次(radius=2)。最终结果在此不做展示。
生成邻近矩阵
邻接矩阵(adjacency matrix)是一种方阵,用来表示有限图。它的每个元素代表各点之间是否有边相连。图的关联矩阵需要和邻接矩阵区分。邻接矩阵的具体可以参考:
https://www.leixue.com/ask/what-is-adjacency-matrix
def create_adjacency(mol):
adjacency = Chem.GetAdjacencyMatrix(mol)
return np.array(adjacency)
根据邻接矩阵的定义,我们可以根据第一行为例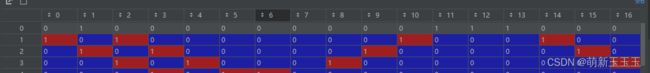
与0号元素相邻的元素有1号,11号,12号和13号。于是在邻接矩阵上就表示为1。有了邻接矩阵,再用了我们之前所编码的信息,就有了矩阵乘法的向量基础。
Sequence向量编码
def split_sequence(sequence, ngram):
sequence = '-' + sequence + '='
# print(sequence)
words = [word_dict[sequence[i:i+ngram]] for i in range(len(sequence)-ngram+1)]
return np.array(words)
这段代码比较好解释,核心就是将sequence的一维序列,每三个字符进行一个编码,但是并非是三个三个拿取,而是依次推进取三个字符。

最终的编码结果为:

至此,数据处理部分就做完了。
总结一下保存的数据为:

compounds:通过“指纹”处理之后的原子编码
adjacencies:邻接矩阵
regression:np.array([math.log2(float(Kcat))])
proteins:words,sequence向量编码
手性添加方案
从数据处理部分我们可以看出,通过化学键和原子的编码可以很好的确定一个小分子的结构。但是在编码上,没有考虑手性的信息。这里我们采用给顺、逆手性的原子进行特殊编码的一个思路。
比如说:
> s='N[C@@](Br)(Cl)OCC[C@@](O)(N)C'
mol=Chem.MolFromSmiles(s)
p=Chem.FindMolChiralCenters(mol)
print(p)
'''-----------------------------------'''
>>>[(1, 'S'), (7, 'R')]
这里的输出一号原子和七号原子为标记为手性的原子,但是其本身依然是C原子,根据原子编码,C原子的编码为0,这里我们令为顺时针手性的C原子编码为一个数值较大的新值,逆时针手性的C原子编码为一个较小的数值。
这样做的目的:突出位点的信息特征,使模型将这个原子作为一个新原子学习到。但是存在两点疑虑。
- 通过这样的编码,等于变相改变了原子的结构,模型会认为你这是一个新的原子,与C原子做了区分。
- 通过“指纹”的循环之后,与较大数值相连的原子数值权重会变得越来越大,而与较小数值相连的数值权重,则没有那么大。
这种编码方式在理论上是有瑕疵的,真正实现是比较好实现的。其实现方式如下:
#手性
max_atoms = max(atoms)
min_atoms = min(atoms)
p = Chem.FindMolChiralCenters(mol)
for i in range(len(p)):
if p[i][1] == 'R':
atoms[p[i][0]] = max_atoms + 1
elif p[i][1] == 'S':
atoms[p[i][0]] = min_atoms - 1