生信人的20个R语言习题
上学期在学校跟练了GEO数据挖掘,看了TCGA数据挖掘有关知识,还没来得及实操。假期摸鱼自学了一下数据挖掘—基于R语言的实践,一直没找找到书本配套实操文件,只能过一遍理论知识,现在跟做Jimmy大神的生信人的20个R语言习题。
原始题目:
生信人的20个R语言习题
参考文章:
20个R语言习题(notion上 还有很多知识可以跟着学)
生信人的20个R语言习题及其答案-土豆学习笔记
R语言20练习题【完整版】
生信人的20个R语言习题(和第一个一样详细)
生信人的20个R语言习题
-
-
- 1.安装R语言包
-
- 1.1R包安装的三种方式
- 1.2安装
- 2.了解ExpressionSet对象
-
- 2.1ExpressionSet相关知识
- 2.2R操作部分
- 3.了解str、head、help、ggplot2函数
-
- 3.1基本知识
- 3.2基本绘图语法
- 4.安装并了解 hgu95av2.db 包
- 5.理解head(toTable(hgu95av2SYMBOL))的用法
- 6.理解探针与基因的对应关系
- 7.找到那些不在 hgu95av2.db 包收录的对应着SYMBOL的探针
- 8.过滤表达矩阵
- 9.整合表达矩阵
- 10.把过滤后的表达矩阵更改行名为基因的symbol
- 11.探索表达矩阵
- 12.统计学指标
- 13.热图可视化矩阵
- 14.不同统计指标的交集
- 15.提取表型数据
- 16.聚类分析
- 17.PCA主成分分析
- 18.批量T检验
- 19.limma
- 20.散点图 火山图
-
1.安装R语言包
不同领域的R包使用频率不一样,在生物信息学领域,尤其需要掌握bioconductor系列包。
数据包:ALL CLL pasilla airway
软件包:limma DESeq2 clusterProfiler
工具包:reshape2
绘图包:ggplot2
1.1R包安装的三种方式
之前一直用的install.packages()的方式安装包,偶有安装不上的老师提示用BiocManager::install()的方式安装,一直不清楚为什么,趁此机会整理一下。
install.packages()函数安装CRAN上的包
CRAN是R综合档案网络的简称,利用install.packages()函数可以单个包作为变量传输进入,也可以通过向量的形式进行多个R包的安装。
#单个包安装
> install.packages("tidyverse")
#多个包的安装
> install.packages(c("tidyverse","ggstatsplot"))
生物信息R包的安装
我们常用的生信R包不在CRAN上,而是在bioconductor上,因此需要用BiocManager::install函数安装,先用基础方式安装BiocManager,安装之前可以利用if函数判断是否已经存在。
> if(!require("BiocManager",quitely=TRUE))
install.packages("BiocManager")
> BiocManager::insrall("DESeq2")
> library(DESeq2)
require()加载包,如果不存在会发出警告信息返回FALSE但据继续执行程序,所以当不存在的时候返回F,!代表非,if判断语句的结果是T就执行install语句安装包。library()加载包,但如果包不存在会报错无法继续执行。
安装非正式版的包
有一些未通过CRAN或bioconductor审核的包存在于github、gitlab中,需要使用devtools或romote包。
具体参考:R包安装教程 | 熟悉的配方但不一样的味道
1.2安装
#修改镜像
> source("http://bioconductor.org/biocLite.R")
> options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
> options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")
#先判断是否存在再安装
> if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
> if (!requireNamespace("ALL", quietly = TRUE))
BiocManager::install("ALL")
> if (!requireNamespace("CLL", quietly = TRUE))
BiocManager::install("CLL")
> if (!requireNamespace("pasilla", quietly = TRUE))
BiocManager::install("pasilla")
> if (!requireNamespace("airway", quietly = TRUE))
BiocManager::install("airway")
> if (!requireNamespace("limma", quietly = TRUE))
BiocManager::install("limma")
> if (!requireNamespace("DESeq2", quietly = TRUE))
BiocManager::install("DESeq2")
> if (!requireNamespace("clusterProfiler", quietly = TRUE))
BiocManager::install("clusterProfiler")
> if (!requireNamespace("reshape2", quietly = TRUE))
BiocManager::install("reshape2")
> if (!requireNamespace("ggplot2", quietly = TRUE))
BiocManager::install("ggplot2")
#加载
> library(ALL)
> library(CLL)
> library(pasilla)
> library(airway)
> library(DESeq2)
> library(clusterProfiler )
> library(reshape2)
> library(ggplot2)
2.了解ExpressionSet对象
比如CLL包里有data(cCLLex),找到它包含的元素,提取其表达矩阵(使用exprs函数),查看其大小。
参考:
- 用limma对芯片数据做差异分析
- ExpressionSet 对象简单讲解
2.1ExpressionSet相关知识
ExpressionSet的组成
ExpressionSet类旨在将多个不同的信息源组合到一个方便的结构中去,是很多Bioconductor函数的输入或输出。
- assayData:微阵列表达数据
- phenodata:实验样本的描述
- featuredata:实验所用芯片或技术的特点
- annotation:注释信息
- experimentData:描述数据的一种灵活结构
2.2R操作部分
CLL数据集是ExpressionSet格式的,包括慢性淋巴细胞白血病(Chroniclymphocytic leukemia)的信息,它采用了Affymetrix公司的HG_U95Av2表达谱芯片(含有12625个探针组),共测量了22个样品(“CLL2.CEL” “CLL3.CEL” “CLL4.CEL” “CLL5.CEL” "CLL6.CEL"等),每个样品来自一个癌症病人,所有病人根据健康状态分为两组:稳定期(Stable)组和进展期(Progressive)也称为恶化期组。
。
0.隐藏具体加载信息 查看CLL包
> suppressPackageStartupMessages(library(CLL))
> ?CLL
#在右下角窗口可以看到CLL包的介绍 了解到读取该包的数据需要data函数
1.读取数据
> data("sCLLex")
> sCLLex
ExpressionSet (storageMode: lockedEnvironment)
assayData: 12625 features, 22 samples
element names: exprs
protocolData: none
phenoData
sampleNames: CLL11.CEL CLL12.CEL ... CLL9.CEL (22 total)
varLabels: SampleID Disease
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation: hgu95av2
#由sCLLex得到的信息可以确定ExpressionSet的组成和该数据集内容和之前说明的一致
2.获得表达矩阵
> exprSet<-exprs(sCLLex)
3.探索表达矩阵
#查看对象结构
> str(exprSet)
num [1:12625, 1:22] 5.74 2.29 3.31 1.09 7.54 ...
- attr(*, "dimnames")=List of 2
..$ : chr [1:12625] "1000_at" "1001_at" "1002_f_at" "1003_s_at" ...
..$ : chr [1:22] "CLL11.CEL" "CLL12.CEL" "CLL13.CEL" "CLL14.CEL" ...
#查看对象前六行
> head(exprSet)
CLL11.CEL CLL12.CEL CLL13.CEL CLL14.CEL CLL15.CEL
1000_at 5.743132 6.219412 5.523328 5.340477 5.229904
1001_at 2.285143 2.291229 2.287986 2.295313 2.662170
1002_f_at 3.309294 3.318466 3.354423 3.327130 3.365113
1003_s_at 1.085264 1.117288 1.084010 1.103217 1.074243
1004_at 7.544884 7.671801 7.474025 7.152482 6.902932
1005_at 5.083793 7.610593 7.631311 6.518594 5.059087
#查看维度
> dim(exprSet)
[1] 12625 22
#查看样本编号
> sampleNames(sCLLex)
[1] "CLL11.CEL" "CLL12.CEL" "CLL13.CEL" "CLL14.CEL" "CLL15.CEL"
[6] "CLL16.CEL" "CLL17.CEL" "CLL18.CEL" "CLL19.CEL" "CLL20.CEL"
[11] "CLL21.CEL" "CLL22.CEL" "CLL23.CEL" "CLL24.CEL" "CLL2.CEL"
[16] "CLL3.CEL" "CLL4.CEL" "CLL5.CEL" "CLL6.CEL" "CLL7.CEL"
[21] "CLL8.CEL" "CLL9.CEL"
#查看所有表型变量
> varMetadata(sCLLex)
labelDescription
SampleID Sample ID
Disease Stable/Progressive
#查看基因编码芯片
> featureNames(sCLLex)[1:10]
[1] "1000_at" "1001_at" "1002_f_at" "1003_s_at" "1004_at"
[6] "1005_at" "1006_at" "1007_s_at" "1008_f_at" "1009_at"
#查看分组信息
> phenoData(sCLLex)
An object of class 'AnnotatedDataFrame'
sampleNames: CLL11.CEL CLL12.CEL ... CLL9.CEL (22 total)
varLabels: SampleID Disease
varMetadata: labelDescription
#查看是否有基因编码重复的
> featureNames(sCLLex) %>% unique() %>% length()
[1] 12625
4.提取表型信息
> pdata<-pData(sCLLex)
> head(pdata)
SampleID Disease
CLL11.CEL CLL11 progres.
CLL12.CEL CLL12 stable
CLL13.CEL CLL13 progres.
CLL14.CEL CLL14 progres.
CLL15.CEL CLL15 progres.
CLL16.CEL CLL16 progres.
#从数据框中提取出表型信息 即只需要知道稳定期还是进展期
> group_list=as.character(pdata$Disease)
#统计表型信息
> table(group_list)
> group_list
progres. stable
14 8
#绘制芯片数据质量图
> y<-melt(as.data.frame(exprSet))
> p<-ggplot(data=y,aes(x=variable,y=value))+
geom_boxplot(aes(fill=variable))+
theme(axis.text.x = element_text(angle = 45,hjust = 1,vjust = 1))+
xlab("分组信息")+ylab("表达值")+guides(fill=FALSE)
p

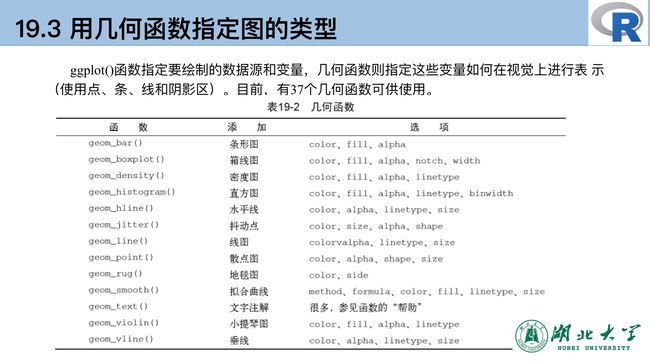
3.了解str、head、help、ggplot2函数
了解上述函数,主要用于第二部提取表达矩阵,因为第二问我已经使用过了,这里就不多说明,这里记载一下ggplot2绘图相关的知识。
3.1基本知识
概念
ggplot2基于图形语法提供一种全新的图形创建方式,绘图过程为数据data、转换transformation、度量scale、坐标系coordinate、元素element、指引guide、显示display 等一系列独立的步骤。通过这些步骤搭配组合,来实现个性化的统计绘图。
图层
- Data 数据: Input data
- Geom 几何对象: A geometry representing data. Points, Lines etc
- Aesthetic 视觉通道: Visual characteristics of the geometry. Size, Color, Shape etc
- Scale 度量: How visual characteristics are converted to display values
- Statistics 统计: Statistical transformations. Counts, Means etc
- Coordinates 坐标: Numeric system to determine position of geometry. Cartesian, Polar etc
- Facets 分面: Split data into subsets
图片和相关知识来自:R语言可视化

3.2基本绘图语法

data为数据集,主要是数据框格式的数据集。
mapping为变量的视觉通道映射,用来表示变量x和y,还可以用来控制颜色、大小、形状等视觉通道。
aes函数指定每个变量扮演的角色,在aes(x=wt,y=mpg)中变量wt的值映射到沿x轴的距离,变量mpg的值映射到沿y轴的距离。
一些函数说明,使用的时候可来查询,也可查询上面链接的文章。






4.安装并了解 hgu95av2.db 包
看看 ls(“package:hgu95av2.db”) 后显示的那些变量,hgu95av2.db是一个注释包,它为hgu95av2平台的芯片提供注释,这个包中有很多注释文件。
这个包里有35个映射数据,都是把芯片探针ID号转换其他主流ID号的映射,每一个数据都是一个包,比如hgu95av2GO就是对应到GO注释,可以得到GO里面的ID号。
参考文章:Bioconductor系列之hgu95av2.db
> BiocManager::install("hgu95av2.db")
> library(hgu95av2.db)
> ls("package:hgu95av2.db")
[1] "hgu95av2" "hgu95av2.db" "hgu95av2_dbconn"
[4] "hgu95av2_dbfile" "hgu95av2_dbInfo" "hgu95av2_dbschema"
[7] "hgu95av2ACCNUM" "hgu95av2ALIAS2PROBE" "hgu95av2CHR"
[10] "hgu95av2CHRLENGTHS" "hgu95av2CHRLOC" "hgu95av2CHRLOCEND"
[13] "hgu95av2ENSEMBL" "hgu95av2ENSEMBL2PROBE" "hgu95av2ENTREZID"
[16] "hgu95av2ENZYME" "hgu95av2ENZYME2PROBE" "hgu95av2GENENAME"
[19] "hgu95av2GO" "hgu95av2GO2ALLPROBES" "hgu95av2GO2PROBE"
[22] "hgu95av2MAP" "hgu95av2MAPCOUNTS" "hgu95av2OMIM"
[25] "hgu95av2ORGANISM" "hgu95av2ORGPKG" "hgu95av2PATH"
[28] "hgu95av2PATH2PROBE" "hgu95av2PFAM" "hgu95av2PMID"
[31] "hgu95av2PMID2PROBE" "hgu95av2PROSITE" "hgu95av2REFSEQ"
[34] "hgu95av2SYMBOL" "hgu95av2UNIPROT"
#如果直接ls 不加packages会报错 至于原因还没找到
#看其他老师的笔记应该是有36个映射数据 现在只有35个 可能是后面包更新了
使用这个包自带函数capture.output(hgu95av2())可以看到这些映射并没有囊括我们标准的hg19版本的2.3万个基因,也就是这个芯片设计的探针只有1.1万个左右。
> capture.output(hgu95av2())
[1] "Quality control information for hgu95av2:"
[2] ""
[3] ""
[4] "This package has the following mappings:"
[5] ""
[6] "hgu95av2ACCNUM has 12625 mapped keys (of 12625 keys)"
[7] "hgu95av2ALIAS2PROBE has 37476 mapped keys (of 143558 keys)"
[8] "hgu95av2CHR has 11683 mapped keys (of 12625 keys)"
[9] "hgu95av2CHRLENGTHS has 595 mapped keys (of 640 keys)"
[10] "hgu95av2CHRLOC has 11637 mapped keys (of 12625 keys)"
[11] "hgu95av2CHRLOCEND has 11637 mapped keys (of 12625 keys)"
[12] "hgu95av2ENSEMBL has 11609 mapped keys (of 12625 keys)"
[13] "hgu95av2ENSEMBL2PROBE has 10016 mapped keys (of 39708 keys)"
[14] "hgu95av2ENTREZID has 11683 mapped keys (of 12625 keys)"
[15] "hgu95av2ENZYME has 2137 mapped keys (of 12625 keys)"
[16] "hgu95av2ENZYME2PROBE has 785 mapped keys (of 975 keys)"
[17] "hgu95av2GENENAME has 11683 mapped keys (of 12625 keys)"
[18] "hgu95av2GO has 11475 mapped keys (of 12625 keys)"
[19] "hgu95av2GO2ALLPROBES has 20621 mapped keys (of 22963 keys)"
[20] "hgu95av2GO2PROBE has 15988 mapped keys (of 18933 keys)"
[21] "hgu95av2MAP has 11681 mapped keys (of 12625 keys)"
[22] "hgu95av2OMIM has 11071 mapped keys (of 12625 keys)"
[23] "hgu95av2PATH has 5445 mapped keys (of 12625 keys)"
[24] "hgu95av2PATH2PROBE has 228 mapped keys (of 229 keys)"
[25] "hgu95av2PMID has 11673 mapped keys (of 12625 keys)"
[26] "hgu95av2PMID2PROBE has 570355 mapped keys (of 754859 keys)"
[27] "hgu95av2REFSEQ has 11646 mapped keys (of 12625 keys)"
[28] "hgu95av2SYMBOL has 11683 mapped keys (of 12625 keys)"
[29] "hgu95av2UNIPROT has 11416 mapped keys (of 12625 keys)"
[30] ""
[31] ""
[32] "Additional Information about this package:"
[33] ""
[34] "DB schema: HUMANCHIP_DB"
[35] "DB schema version: 2.1"
[36] "Organism: Homo sapiens"
[37] "Date for NCBI data: 2021-Apr14"
[38] "Date for GO data: 2021-02-01"
[39] "Date for KEGG data: 2011-Mar15"
[40] "Date for Golden Path data: 2021-Feb16"
[41] "Date for Ensembl data: 2021-Feb16"
这个hgu95av2.db所加载的36个包都是一种特殊的对象,但是可以把它当做list来操作,是一种类似于hash的对应表格,其中keys是独特的,但是value可以有多个。既然是类似于list,那我就简单讲几个小技巧来操作这些数据对象。所有的操作都要用as.list()函数来把数据展现出来。
> as.list(hgu95av2ENZYME[1])
$`1000_at`
[1] "2.7.11.24"
#可以看到这样就提取出来了hgu95av2ENZYME的第一个元素 key是`1000_at` 它所映射的value是 "2.7.11.24"这个酶
同理对list取元素的三个操作在这里都可以用
> as.list(hgu95av2ENZYME['1000_at'])
> as.list(hgu95av2ENZYME$'1000_at')
#GO注释一般会存在多个 可以用ToTable来格式化
> as.list(hgu95av2GO$'1000_at')
> toTable(hgu95av2GO[1])
5.理解head(toTable(hgu95av2SYMBOL))的用法
理解head(toTable(hgu95av2SYMBOL))的用法,即先把hgu95av2SYMBOL对象表格化(便于查看),然后展示前六行,可以看到列名是probe_id和symbol,前者是探针ID,后者是基因名,一个探针对应一个基因名,即描述了厂家的标记符—探针和基因缩写之间的映射关系。Jimmy大神让找到TP53 基因对应的探针ID。
关于基因探针部分的知识参考:GEO数据库学习一(简介 数据下载 芯片知识)
toTable
#是一种能够以数据框的形式来操作一个Bimap对象的方法 也就是把Bimap对象转换为一个数据框
#Bimap指的是一种映射关系 例如探针的编号与基因名称之间的映射
#查看hgu95av2SYMBOL包
#Lkeys:探针总数12625 实际匹配上的11683 指有基因与其对应的探针
#Rkeys:基因名的总数77510 实际上只有8776
#原因是一个基因可以对应多个探针 取最大表达值的探针作为基因的表达量
> summary(hgu95av2SYMBOL)
SYMBOL map for chip hgu95av2 (object of class "ProbeAnnDbBimap")
|
| Lkeyname: probe_id (Ltablename: probes)
| Lkeys: "1000_at", "1001_at", ... (total=12625/mapped=11683)
|
| Rkeyname: symbol (Rtablename: gene_info)
| Rkeys: "A1BG", "A2M", ... (total=77510/mapped=8776)
|
| direction: L --> R
#将其表格化
> gene_id<-toTable(hgu95av2SYMBOL)
> head(gene_id)
probe_id symbol
1 1000_at MAPK3
2 1001_at TIE1
3 1002_f_at CYP2C19
4 1003_s_at CXCR5
5 1004_at CXCR5
6 1005_at DUSP1
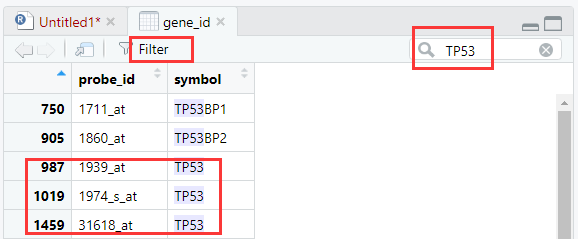
#查看TP53对应的探针
#filter过滤 括号里面前者是数据对象 后者是筛选条件
> filter(gene_id,symbol=="TP53")
probe_id symbol
1 1939_at TP53
2 1974_s_at TP53
3 31618_at TP53
#grep正则表达式 ^锚定开头为TP53的 $结尾 grep抓取符合条件的行 其余列一起列出
> gene_id[grep("^TP53$",gene_id$symbol),]
probe_id symbol
987 1939_at TP53
1019 1974_s_at TP53
1459 31618_at TP53
其实也可以View(gene_id)后直接在搜索框搜,这种适用于仅需查看不需要后续操作,可以作为一种检验方法。
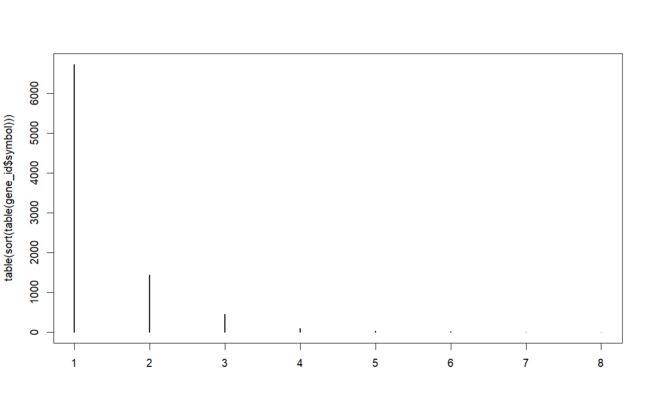
6.理解探针与基因的对应关系
总共多少个基因,基因最多对应多少个探针,是哪些基因,是不是因为这些基因很长,所以在其上面设计多个探针呢?
不管是Agilent芯片,还是Affymetrix芯片,上面设计的探针都非常短。最长的如Agilent芯片上的探针,往往都是60bp,但是往往一个基因的长度都好几Kb。因此一般多个探针对应一个基因,取最大表达值探针来作为基因的表达量。
#查看有多少基因 unique去除重复
> length(unique(gene_id$symbol))
[1] 8776
#查看最后几个基因涉及到的探针数目
#table()就是统计每一种的个数 生成一个列表 它是对个数的统计
#第一次table统计基因名出自的个数 得到列表的数字代表这种基因出现了几次 也就是这种基因对应的探针数目
#第二次table统计的就是上一次统计出来的数字出现的个数 也就是某一种基因的探针个数 从结果可以知道大多数基因都是一个探针 少数有多个探针
> tail(sort(table(gene_id$symbol)))
RBPMS TERF1 INPP4A MYB PTGER3 STAT1
7 7 8 8 8 8
#查看所有基因涉及到的探针数目
> table(sort(table(gene_id$symbol)))
1 2 3 4 5 6 7 8
6725 1444 451 103 27 16 6 4
#画图
> plot(table(sort(table(gene_id$symbol))))
7.找到那些不在 hgu95av2.db 包收录的对应着SYMBOL的探针
第二步提取到的表达矩阵是12625个探针在22个样本的表达量矩阵,找到那些不在 hgu95av2.db 包收录的对应着SYMBOL的探针。
A提示:有1165(942)个探针是没有对应基因名字的。
第二步得到的表达矩阵exprSet是12625个探针,按理说这些探针在hgu95av2.db中都能找到相匹配的symbol,但有些基因可能没有收录在这个包里,现在需要找出他们并将其去除。即最后留下有对应关系的那些基因。基本思路是利用hgu95av2SYMBOL中探针和symbol的对应关系,找到hgu95av2SYMBOL包中能够与基因映射起来的探针,再找到这些探针对应的基因SYMBOL,然后在exprSet中取出这些基因即可,这一步主要是取出不存在的那些基因。
mappedkeys()映射关系
#求全部探针数目长度并赋给probe_map
> length(hgu95av2SYMBOL)
[1] 12625
> probe_map<-hgu95av2SYMBOL
> length(probe_map)
[1] 12625
#探针与基因产生映射的数目
#mappedkeys的作用是将hgu95av2SYMBOL中有映射关系的取出来
> probe_info<-mappedkeys(probe_map)
> length(probe_info)
[1] 11683
#转化为数据表
> gene_info<-as.list(probe_map[probe_info])
#从hgu95av2SYMBOL文件中 取出有映射关系的探针 并生成数据框
> gene_symbol<-toTable(probe_map[probe_info])
> head(gene_symbol)
probe_id symbol
1 1000_at MAPK3
2 1001_at TIE1
3 1002_f_at CYP2C19
4 1003_s_at CXCR5
5 1004_at CXCR5
6 1005_at DUSP1
#在exprSet中挑出gene_symbol
> filter_exprSet<-exprSet[rownames(exprSet) %in% gene_symbol$probe_id,]
> unfilter_exprSet<-exprSet[!(rownames(exprSet) %in% gene_symbol$probe_id),]
> head(filter_exprSet)
mappedkeys()函数用于处理映射文件(bimap) 参数是Bimap对象
%in%逻辑判断
这里直接让exprSet的探针与hgu95av2SYMBOL的探针进行匹配,匹配到11683个,刚好就是有探针和基因名对应关系的11683,至于原因还没想明白。
a %in% b
逻辑判断a是否存在于b中 如果存在结果为T 如果不存在结果为F
#查看exprSet中的探针有多少在hgu95av2.db
#这里只有942个 可能是因为包更新
> table(rownames(exprSet) %in% gene_id$probe_id)
FALSE TRUE
942 11683
#将不对应的探针提取出来
> new_exprSet<-exprSet[!(rownames(exprSet) %in% gene_id$probe_id),]
> dim(new_exprSet)
[1] 942 22
8.过滤表达矩阵
过滤表达矩阵,删除那942个没有对应基因名字的探针。这里基本就是上一题的收尾,两题可以合在一起,这两部分的代码基本一样。
方法一:%in%逻辑判断
#过滤表达矩阵 删除942个没有对应基因名字的探针
> exprSet<-exprSet[rownames(exprSet) %in% gene_id$probe_id,]
> dim(exprSet)
[1] 11683 22
方法二:mappedkeys()映射关系
#求全部探针数目长度并赋给probe_map
> length(hgu95av2SYMBOL)
[1] 12625
> probe_map<-hgu95av2SYMBOL
> length(probe_map)
[1] 12625
#探针与基因产生映射的数目
#mappedkeys的作用是将hgu95av2SYMBOL中有映射关系的取出来
> probe_info<-mappedkeys(probe_map)
> length(probe_info)
[1] 11683
#转化为数据表
> gene_info<-as.list(probe_map[probe_info])
#从hgu95av2SYMBOL文件中 取出有映射关系的探针 并生成数据框
> gene_symbol<-toTable(probe_map[probe_info])
> head(gene_symbol)
probe_id symbol
1 1000_at MAPK3
2 1001_at TIE1
3 1002_f_at CYP2C19
4 1003_s_at CXCR5
5 1004_at CXCR5
6 1005_at DUSP1
#在exprSet中挑出gene_symbol
> filter_exprSet<-exprSet[rownames(exprSet) %in% gene_symbol$probe_id,]
mappedkeys()函数用于处理映射文件(bimap) 参数是Bimap对象
9.整合表达矩阵
整合表达矩阵,多个探针对应一个基因的情况下,只保留在所有样本里面平均表达量最大的那个探针。
A.提示,理解 tapply,by,aggregate,split 函数 , 首先对每个基因找到最大表达量的探针;
B.然后根据得到探针去过滤原始表达矩阵。
基本思路:gene_symbol是已经过滤的探针与symbol对应的数据框,filter_exprSet是已经过滤好的表达矩阵(即这里面的探针都有symbol与其对应)(表达矩阵行是探针 列名是样品名 值是表达量),即沿利用探针将基因名与基因表达量对应起来,对于多个探针对应同一个基因的情况,计算平均表达量,取最大的那一个。
tapply(X,INDEX,FUN)
该函数为向量中的每个因子变量计算一个度量值(均值、中值、最小值、最大值)或一个函数 即可以对每个因子应用一些函数
INDEX:包含因子的列表
apply(X,MARGIN,FUN)
该函数以数据帧或矩阵作为输入 并以向量 列表或数组的形式给出输出 用于遍历数组中的行或列 并且使用指定函数对其处理
X:数组或矩阵
MARGIN:1对行执行 2对列执行 c(1,2)对行和列同时执行
FUN:应用的函数
by(data,INDICES,FUN...,simplify=TRUE)
该函数用于将data中的数据按照INDICES里面的内容拆分成若干小的data frame 并且在每一小块data frame上应用FUN函数
aggregate(X,by,Fun,simplify=TRUE,,drop=TRUE)
该函数将数据拆分成子集 为每个子集计算摘要统计信息 然后以方便的形式返回结果 即按照by指定的列对指定的X对象分组 同时对X作Fun运算
X:R对象
by:分组元素的列表 每个与数据框X中的变量长度相同 使用之前 将这些元素强制转换为因子 即这是分组的依据
Fun:计算汇总的函数
simplify:表明是否将结果简化为矩阵或向量
drop:表明是否删除未使用分组值组合的逻辑值 默认T
match()
该函数返回第一个元素在第二个参数中的第一个匹配位置的向量
1.将表达矩阵filter_exprSet中的探针名称排序
#order()返回的值是原有行向量的位置 然后数据框取对应位置的值
> filter_exprSet_order<-filter_exprSet[order(rownames(filter_exprSet)),]
2.对gene_symbol按照探针名称进行排序
> gene_symbol_order<-gene_symbol[order(gene_symbol$probe_id),]
#查看地表达值的矩阵的名称是否与gene_symbol中的探针名称是否一一对应
> table(rownames(filter_exprSet_order)==gene_symbol_order$probe_id)
TRUE
11683
3.计算每一行的平均值
> row_mean<-apply(filter_exprSet_order,1,mean)
> head(row_mean)
100_g_at 1000_at 1001_at 1002_f_at 1003_s_at 1004_at
2.289762 5.456561 2.334098 3.402072 1.126168 7.439034
4.保留相同基因中表达量最高的那一行
#构建函数 挑出最大的一个
> index_probe<-function(x){
+ names(x)[which.max(x)]
+ }
#index_probe这个函数用gene_symbol$symol(基因名)对row_mean进行分组 然后挑出相同基因中表达值最大的那个值所对应的探针编号
> max_index<-tapply(row_mean,gene_symbol$symbol,index_probe)
> head(max_index)
AADAC AAK1 AAMP AANAT AARS1 AASDHPPT
"36511_at" "40571_at" "38433_at" "36331_at" "36184_at" "35760_at"
#挑出表达量最大值
> filter_exprSet_order_unique<-filter_exprSet_order[gene_symbol_order$probe_id %in% max_index,]
#将filter_exprSet_order_unique那些与gene_symbol_order$probe_id对应起来的探针与基因名提取出来
> unique_map_gene_probe <- gene_symbol_order[(gene_symbol_order$probe_id %in% rownames(filter_exprSet_order_unique)),]
#统计筛选后的基因总数 这个数值应该和table(sort(table(gene_id$symbol)))得到的数字相加结果一样
> table(unique_map_gene_probe$probe_id == rownames(filter_exprSet_order_unique))
TRUE
8776
10.把过滤后的表达矩阵更改行名为基因的symbol
#把过滤后的表达矩阵更改行名为基因的symbol
rownames(filter_exprSet_order_unique)<-unique_map_gene_probe$symbol
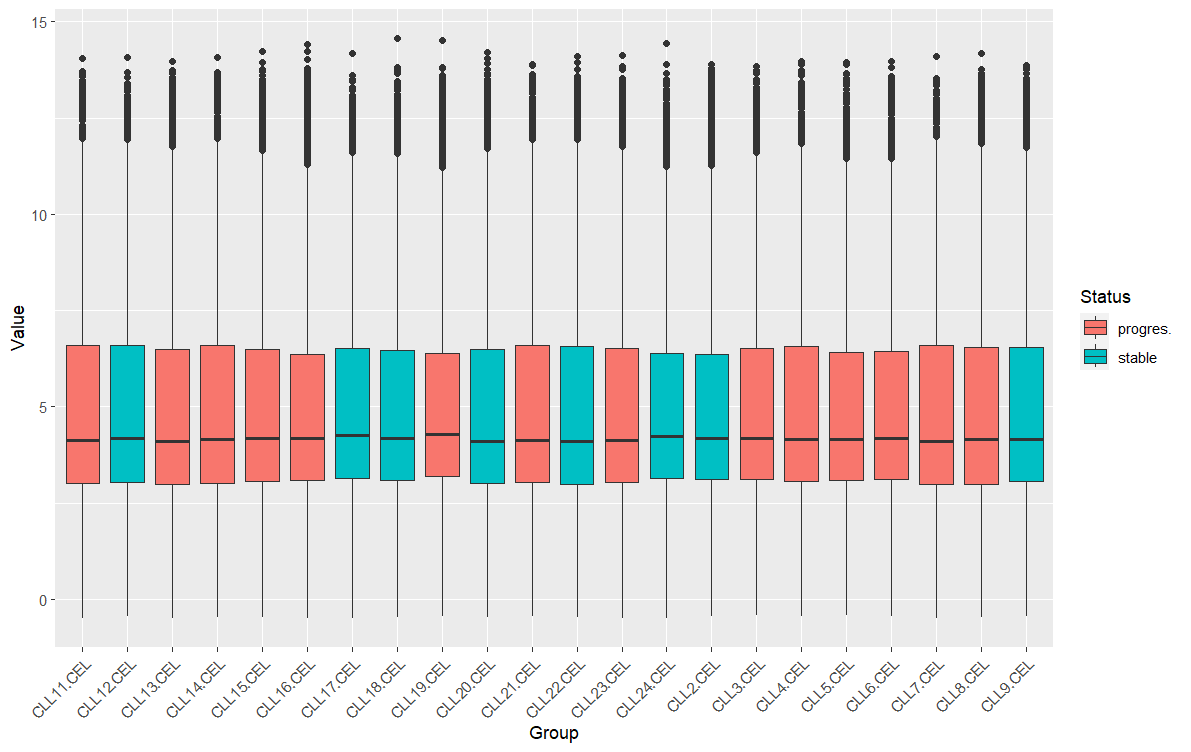
11.探索表达矩阵
对第10步得到的表达矩阵进行探索,先画第一个样本的所有基因的表达量的boxplot,hist,density,然后画所有样本的这些图
A.参考:basic visualization for expression matrix;
B.理解ggplot2的绘图语法,数据和图形元素的映射关系。
使用ggplot2绘图,要把数据转换为ggplot2可识别的形式,即第一列基因名,第二列分组信息,第三列表达值。
整理数据形式
#加载包
> library(ggplot2)
> library(reshape2)
#melt()函数对filter_exprSet_order_unique数据库进行拆分
> data_draw<-melt(filter_exprSet_order_unique)
> View(data_draw)
#修改列名
> colnames(data_draw)<-c("Gene","Group","Value")
#添加状态列
#pdata$Disease来自第二问 可以去前面查看
> data_draw$Status<-rep(pdata$Disease,each=nrow(filter_exprSet_order_unique))
图像部分只有代码和图形,具体说明见下次系统学习完后补充。
箱线图
#看数据分布 横轴是组别 纵轴是表达量 填充是状态
#一个样本
> ggplot(data_draw[data_draw[,2]=="CLL11.CEL",],aes(x=Group,y=Value,fill=Status))+geom_boxplot()
#所有样本
> ggplot(data=data_draw,aes(x=Group,y=Value,fill=Status))+geom_boxplot()+theme(axis.title.x = element_text(angle = 45,hjust = 1,vjust = 1))
小提琴图
#看数据分布 和箱线图一致
> ggplot(data = data_draw,aes(x=Group,y=Value,fill=Status)+geom_violin()+theme(axis.text.x = element_text(angle=45,hjust=1,vjust=1))

直方图
#局部或整体 横轴表达量 纵轴统计个数
#facet_wrap(~Group,nrow=5)刻面 将样本按照Group每行5个进行分组绘制 每个样本都有单独的一个图 如果不加就是一个图
> ggplot(data=data_draw,aes(x=Value,fill=Status))+geom_histogram(bins=500)
> ggplot(data=data_draw,aes(x=Value,fill=Status))+geom_histogram(bins=500)+facet_wrap(~Group,nrow=5)


密度图
> ggplot(data=data_draw,aes(x=Value,fill=Status))+geom_density()+facet_wrap(~Group,nrow=5)

12.统计学指标
理解统计学指标mean,median,max,min,sd,var,mad并计算出每个基因在所有样本的这些统计学指标,最后按照mad值排序,取top 50 mad值的基因,得到列表。
mad
mad是绝对中位数差
基本用法:mad(x, center = median(x), constant = 1.4826, na.rm = FALSE, low = FALSE, high = FALSE)
x:数值向量
center:可选 是中心点 默认是中位数
constant:是缩放因子(scale factor) na.rm如果是TRUE 在计算前将x中的NA删除 默认1.4826
low:如果是TRUE 计算lo-median 也就是说 对于个数为偶数的样本 在最后计算中位数时不使用两个中间值的均值 而是采用其中较小的值
high:如果为TRUE 计算‘hi-median’ 也就是对于偶数样本采用两个中间值的较大者作为中位数
#计算mean median max min sd var mad
> g_mean<-tail(sort(apply(filter_exprSet_order_unique,1,mean)),50)
> g_median<-tail(sort(apply(filter_exprSet_order_unique,1,median)),50)
> g_max<-tail(sort(apply(filter_exprSet_order_unique,1,max)),50)
> g_min<-tail(sort(apply(filter_exprSet_order_unique,1,min)),50)
> g_sd<-tail(sort(apply(filter_exprSet_order_unique,1,sd)),50)
> g_var<-tail(sort(apply(filter_exprSet_order_unique,1,var)),50)
> g_mad<-tail(sort(apply(filter_exprSet_order_unique,1,mad)),50)
> names(g_mad)
[1] "PFN2" "CLASRP" "PFN2" "TNFSF9" "ARHGAP44" "P2RY14"
[7] "THEMIS2" "LPL" "ANXA4" "DUSP6" "DUSP5" "IGFBP4"
[13] "H1-10" "CLEC2B" "ZNF266" "S100A9" "NR4A2" "TGFBI"
[19] "ARF6" "APBB2" "PDE4DIP" "CAPG" "RGS2" "RNASE6"
[25] "VAMP5" "CYBB" "GNLY" "CCL3" "OAS1" "ZNF804A"
[31] "H1-10" "IGH" "JUND" "SLC25A1" "PCDH9" "VIPR1"
[37] "COBLL1" "GUSBP11" "S100A8" "HBB" "LHFPL2" "FCN1"
[43] "ZAP70" "LGALS1" "HBB" "FOS" "TCF7" "DMD"
[49] "IGF2BP3" "FAM30A"
13.热图可视化矩阵
根据第12步骤得到top 50 mad值的基因列表来取表达矩阵的子集,并且热图可视化子表达矩阵。试试看其它5种热图的包的不同效果。
heatmap
#通常我们要查看某个基因在不同样本中的表达情况 就采用row进行均一化
#上面的这个数据行是基因名 列是样本名 因此采用行均一化,就可以看相同的某个基因在不同样本中的表达情况
#如果要想看一个样本中不同基因的表达情况 就要采用column进行均一化
> library(pheatmap)
> choose_gene=names(g_mad)
> choose_matrix=filter_exprSet_order_unique[choose_gene,]
> heatmap(choose_matrix,scale = c("row"))
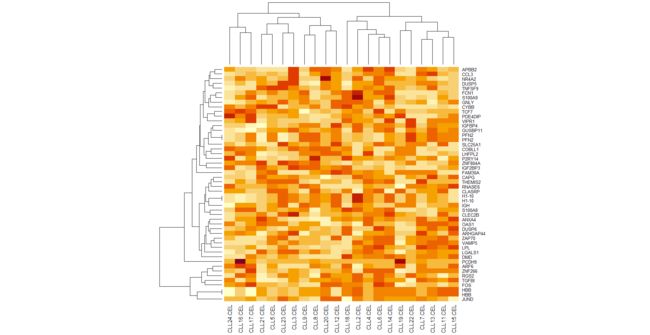
heatmap.2
library(gplots)
heatmap.2(choose_matrix,scale=c("row"))

d3heatmap
d3heatmap包可用于生成交互式热图绘制。
> if (!require("devtools"))
install.packages("devtools")
> devtools::install_github("rstudio/d3heatmap")
> library(d3heatmap)
> d3heatmap(choose_matrix, colors = "RdBu", k_row = 4, k_col = 2)
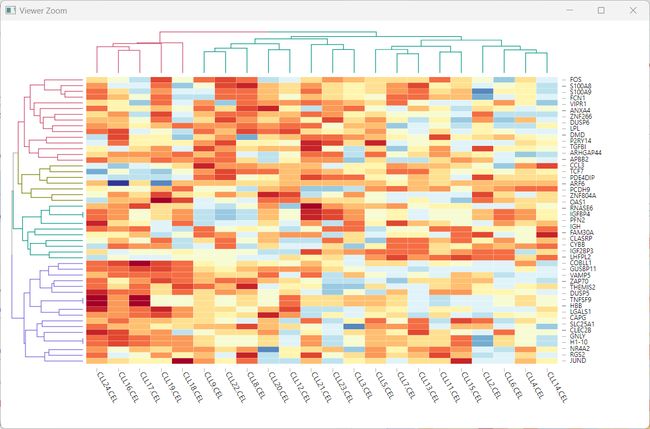
Heatmap
> install.packages("ComplexHeatmap")
> BiocManager::install("ComplexHeatmap")
> library(ComplexHeatmap)
> Heatmap(choose_matrix,name="Mad 30")
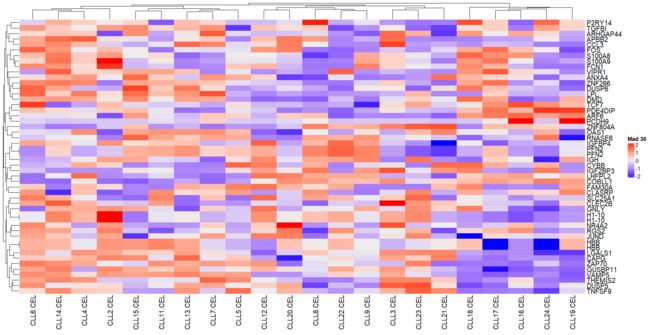
pheatmap
> library(pheatmap)
> pheatmap(data_mad_30_heatmap)
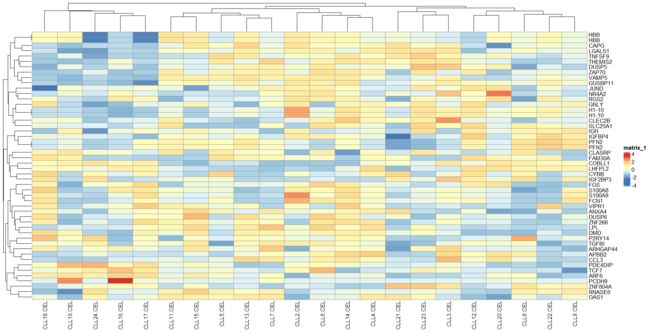
14.不同统计指标的交集
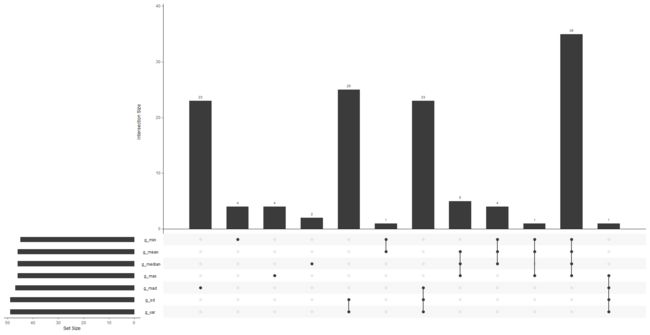
取不同统计学指标mean,median,max,mean,sd,var,mad的各top50基因列表,使用UpSetR包来看他们之间的overlap情况。
#使用UpSetR包看不同统计学指标的重叠程度
> install.packages("UpSetR")
> library(UpSetR)
#取并集
> g_all<-unique(c(names(g_mean),names(g_median),names(g_max),names(g_min),names(g_sd),names (g_var),names(g_mad) ))
#生成数据框 在为1 不在为0
> Data=data.frame(g_all,
g_mean=ifelse(g_all %in% names(g_mean) ,1,0),
g_median=ifelse(g_all %in% names(g_median) ,1,0),
g_max=ifelse(g_all %in% names(g_max) ,1,0),
g_min=ifelse(g_all %in% names(g_min) ,1,0),
g_sd=ifelse(g_all %in% names(g_sd) ,1,0),
g_var=ifelse(g_all %in% names(g_var) ,1,0),
g_mad=ifelse(g_all %in% names(g_mad) ,1,0)
)
> upset(Data,nsets=7)
15.提取表型数据
在第二步的基础上面提取CLL包里面的data(sCLLex) 数据对象的样本的表型数据。
#最原始表达数据
> dim(exprSet)
[1] 11683 22
#筛选过后的 即探针和symbol一一对应的部分
> dim(filter_exprSet_order_unique)
[1] 8776 22
> filter_exprSet_order_unique[1:5,1:5]
CLL11.CEL CLL12.CEL CLL13.CEL CLL14.CEL CLL15.CEL
RABGGTA 2.216194 2.228330 2.223384 2.226454 2.206269
MAPK3 5.743132 6.219412 5.523328 5.340477 5.229904
CYP2C19 3.309294 3.318466 3.354423 3.327130 3.365113
CXCR5 7.544884 7.671801 7.474025 7.152482 6.902932
DUSP1 5.083793 7.610593 7.631311 6.518594 5.059087
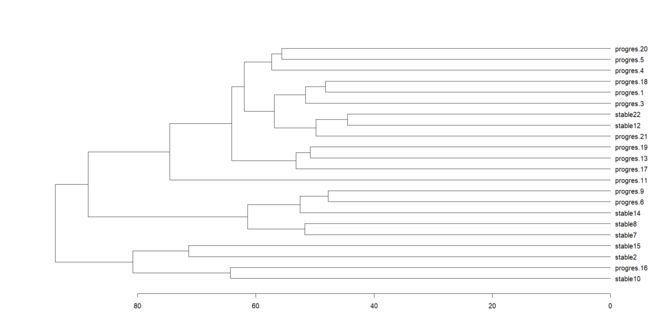
16.聚类分析
对所有样本的表达矩阵进行聚类并且绘图,然后添加样本的临床表型数据信息(更改样本名)。
#更改样本名 更改为样本临床表型信息的样式
colnames(exprSet)=paste(group_list,1:22,sep='')
#定义nodePar
nodepar=list(lab.cex=0.6,pch=c(NA,19),
cex=0.7,col='red')
#聚类
hc=hclust(dist(t(exprSet)))
par(mar=c(5,5,5,10))
#绘图
plot(as.dendrogram(hc),nodepar=nodepar,horiz=TRUE)
17.PCA主成分分析
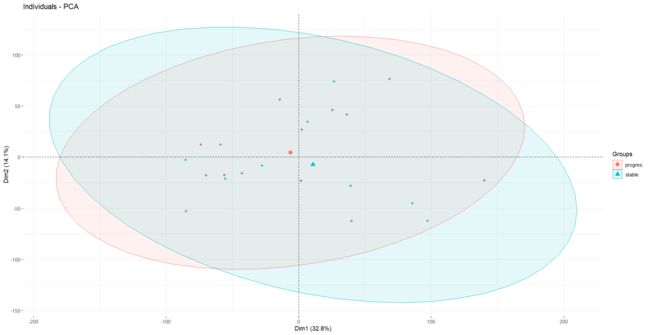
对所有样本的表达矩阵进行PCA分析并且绘图,同样要添加表型信息。
> library("FactoMineR")
> library("factoextra")
> df=as.data.frame(t(exprSet))
> dat.pca <- PCA(df, graph = FALSE)
> fviz_pca_ind(dat.pca,
geom.ind = "point",
col.ind = group_list,
addEllipses = TRUE,
legend.title = "Groups"
)
18.批量T检验
根据表达矩阵及样本分组信息进行批量T检验,得到检验结果表格。
#将表型数据因子化
> group_list
[1] "progres." "stable" "progres." "progres." "progres." "progres." "stable" "stable"
[9] "progres." "stable" "progres." "stable" "progres." "stable" "stable" "progres."
[17] "progres." "progres." "progres." "progres." "progres." "stable"
> gl=as.factor(group_list)
> gl
[1] progres. stable progres. progres. progres. progres. stable stable progres. stable
[11] progres. stable progres. stable stable progres. progres. progres. progres. progres.
[21] progres. stable
Levels: progres. stable
#获取progres的索引
> group1=which(group_list == levels(gl)[1])
#获取stable索引
> group2=which(group_list == levels(gl)[2])
#将表型为progres和stable的样本分别选出来 exprSet是筛选942个没有对应关系的 exprset是原始12625
> data_t1=exprset[,group1]
> data_t2=exprset[,group2]
> data_t=cbind(data_t1,data_t2)
> pvals = apply(exprset, 1, function(x){
+ t.test(as.numeric(x)~group_list)$p.value
+ })
> p.adj = p.adjust(pvals, method = "BH")
#progres对照组 stable处理组
> data_mean_1 = rowMeans(data_t1)
> data_mean_2 = rowMeans(data_t2)
> log2FC = data_mean_2-data_mean_1
> DEG_t.test = cbind(data_mean_1, data_mean_2, log2FC, pvals, p.adj)
> DEG_t.test=DEG_t.test[order(DEG_t.test[,4]),]
> DEG_t.test=as.data.frame(DEG_t.test)
> head(DEG_t.test)
data_mean_1 data_mean_2 log2FC pvals p.adj
36129_at 7.875615 8.791753 0.9161377 1.629755e-05 0.2057566
37676_at 6.622749 7.965007 1.3422581 4.058944e-05 0.2436177
33791_at 7.616197 5.786041 -1.8301554 6.965416e-05 0.2436177
39967_at 4.456446 2.152471 -2.3039752 8.993339e-05 0.2436177
34594_at 5.988866 7.058738 1.0698718 9.648226e-05 0.2436177
32198_at 4.157971 3.407405 -0.7505660 2.454557e-04 0.3516678
19.limma
使用limma包对表达矩阵及样本分组信息进行差异分析,得到差异分析表格,重点看logFC和P值,画个火山图(就是logFC和-log10(P值)的散点图)。
> library(limma)
#这是一个设计矩阵
> design1=model.matrix(~factor(group_list))
> colnames(design1)=levels(factor(group_list))
> rownames(design1)=colnames(exprset)
> fit1 = lmFit(exprset,design1)
> fit1=eBayes(fit1)
#设置结果的小数位数为3
> options(digits = 3)
#coef要么必须等于2 要么是个字符串 关于adjust的设置,说明书中13.1章有描述 BH是最流行的设置
#topTable默认显示前10个基因的统计数据 使用选项n可以设置 n=Inf就是不设上限全部输出
> mtx1 = topTable(fit1,coef=2,adjust='BH',n=Inf)
#去除缺失值
> DEG_mtx1 = na.omit(mtx1)
> head(DEG_mtx1)
logFC AveExpr t P.Value adj.P.Val B
39400_at 1.028 5.62 5.84 8.34e-06 0.0334 3.23
36131_at -0.989 9.95 -5.77 9.67e-06 0.0334 3.12
33791_at -1.830 6.95 -5.74 1.05e-05 0.0334 3.05
1303_at 1.384 4.46 5.73 1.06e-05 0.0334 3.04
36122_at -0.780 7.26 -5.14 4.21e-05 0.1062 1.93
36939_at -2.547 6.92 -5.04 5.36e-05 0.1128 1.74
20.散点图 火山图
对T检验结果的P值和limma包差异分析的P值画散点图,看看哪些基因相差很大。
> DEG=DEG_mtx1
> logFC_cutoff <- with(DEG,mean(abs(logFC)) + 2*sd(abs(logFC)) )
> DEG$result = as.factor(ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT'))
> this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG[DEG$result =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$result =='DOWN',])
)
> ggplot(data=DEG, aes(x=logFC, y=-log10(P.Value), color=result)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red'))
