Jimmy老师春节大放送-GSE190856—TREM2hi resident macrophages protect theseptic heart by maintaining cardiomy
TREM2hi resident macrophages protect the
septic heart by maintaining cardiomyocyte
homeostasis
文章概览


29,537 cardiac immune cells allocated into 15 clusters from the hearts of 14 WT mice at SS, 3, 7 and 21 d after CLP.
本文样本组成比较复杂
我推测
| GSM5733020 | H001 WT non_control |
| GSM5733021 | H002 WT3d |
| GSM5733022 | H003 WT7d |
| GSM5733023 | H004 WT21d |
| GSM5733024 | H005 TREM2 WT 0D |
| GSM5733025 | H006 TREM2 -/- 0D |
| GSM5733026 | H007 TREM2 WT 3D |
| GSM5733027 | H008 TREM2 -/- 3D |
| GSM5733028 | H009 TREM2 WT 7D |
| GSM5733029 | H011 TREM2 -/- 7D |
正式开始之前注意:
#查看分配的内存大小
memory.limit()
#增大分配的内存
memory.limit(size = 3500000)
# Increase limit
就可以防止这种报错

step0:导入数据
###### step1:导入数据 ######
# 付费环节 800 元人民币
dir='GSE190856_RAW'
samples=list.files( dir )
samples
# samples = head(samples,10)
sceList = lapply(samples,function(pro){
# pro=samples[1]
print(pro)
sce =CreateSeuratObject(counts = Read10X(file.path(dir,pro )) ,
project = gsub('^GSM[0-9]*_','',pro) ,
min.cells = 5,
min.features = 500 )
#names(sce)= gsub('^GSM[0-9]*_','',pro)
return(sce)
})
names(sceList)
# gsub('^GSM[0-9]*','',samples)
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ],
add.cell.ids = gsub('_gene_cell_exprs_table.txt.gz','',gsub('^GSM[0-9]*_','',samples) ) )
as.data.frame(sce.all@assays$RNA@counts[1:10, 1:2])
head([email protected], 10)
table(sce.all$orig.ident)
step 1 分组信息
# 分组信息,通常是隐含在文件名,样品名字里面
# phe=str_split( colnames(sce.all),'[-_]',simplify = T)
# head(phe)
# table(phe[,1])
# sce.all$group=toupper( substring(colnames(sce.all),1,2))
# [email protected]$group = ifelse(grepl('ne',sce.all$orig.ident),'ne','e')
# sce.all$group= gsub( '^[0-9]*_' ,'',sce.all$orig.ident)
#sce.all$group = sce.all$orig.ident
table(grepl(pattern = "009",colnames(sce.all)))
table(grepl(pattern = "H001",colnames(sce.all)))
#分组
sce.all$group=ifelse(grepl(pattern = "H001",colnames(sce.all))," WT_non_control",
ifelse(grepl(pattern = "002",colnames(sce.all)),"WT3d",
ifelse(grepl(pattern = "003",colnames(sce.all)),"WT7d",
ifelse(grepl(pattern = "004",colnames(sce.all)),"WT21d",
ifelse(grepl(pattern = "005",colnames(sce.all)),"TREM2_WT_0D",
ifelse(grepl(pattern = "006",colnames(sce.all)),"TREM2_-/-_0D",
ifelse(grepl(pattern = "007",colnames(sce.all)),"TREM2_WT_3D",
ifelse(grepl(pattern = "008",colnames(sce.all)),"TREM2_-/-_3D",
ifelse(grepl(pattern = "009",colnames(sce.all)),"TREM2_WT_7D",
"TREM2_-/-_7D") )))) ) ) ) )
table([email protected]$group)
table([email protected]$orig.ident)

step2:QC质控
###### step2:QC质控 ######
dir.create("./1-QC")
setwd("./1-QC")
getwd()#"G:/silicosis/jimmy_task_study/2023_jimmy_scrna/00-现在完成的/gse190856_septic_heart_macrophage/1-QC"
# 如果过滤的太狠,就需要去修改这个过滤代码
source('../scRNA_scripts/qc.R')
sce.all.filt = basic_qc(sce.all)
print(dim(sce.all))
print(dim(sce.all.filt))
setwd('../')

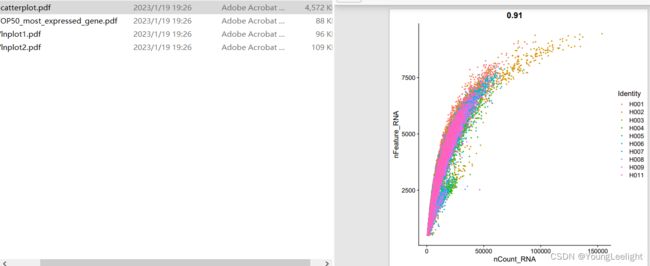
step3: harmony整合多个单细胞样品
sp='mouse'
###### step3: harmony整合多个单细胞样品 ######
dir.create("2-harmony")
getwd()
setwd("2-harmony")
source('../scRNA_scripts/harmony.R')
# 默认 ScaleData 没有添加"nCount_RNA", "nFeature_RNA"
# 默认的
sce.all.int = run_harmony(sce.all.filt)
setwd('../')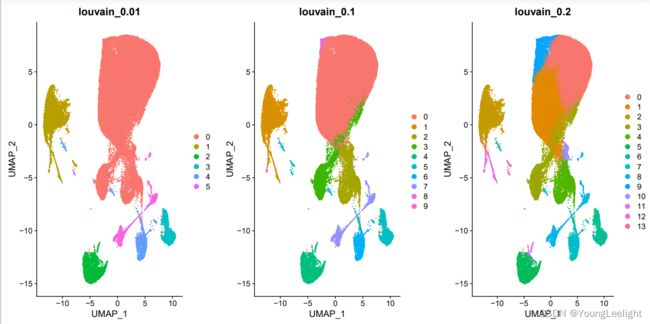
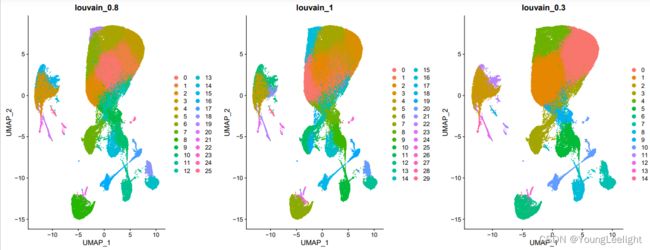

这样看图的话,选择0.1分辨率就可
然后运行下面的代码看看0.1的效果
step4: 降维聚类分群和看标记基因库 ##
step4: 降维聚类分群和看标记基因库 ######
# 原则上分辨率是需要自己肉眼判断,取决于个人经验
# 为了省力,我们直接看 0.1和0.8即可
table(Idents(sce.all.int))
table(sce.all.int$seurat_clusters)
table(sce.all.int$RNA_snn_res.0.1)
table(sce.all.int$RNA_snn_res.0.8)
getwd()
dir.create('check-by-0.1')
setwd('check-by-0.1')
sel.clust = "RNA_snn_res.0.1"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table([email protected])
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
dir.create('check-by-0.1')
setwd('check-by-0.1')
sel.clust = "RNA_snn_res.0.1"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table([email protected])
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
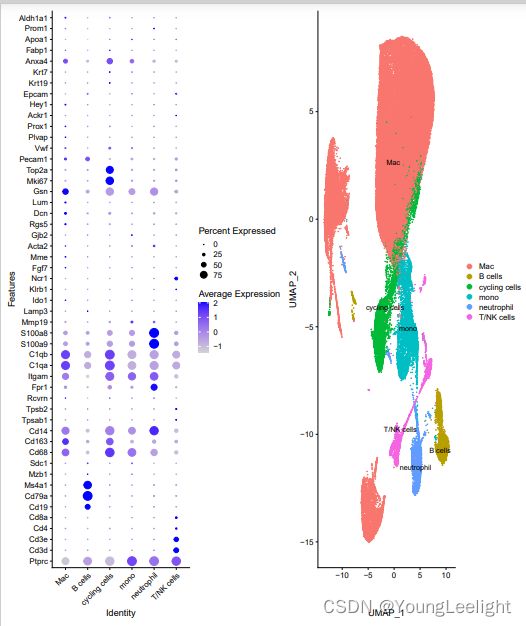
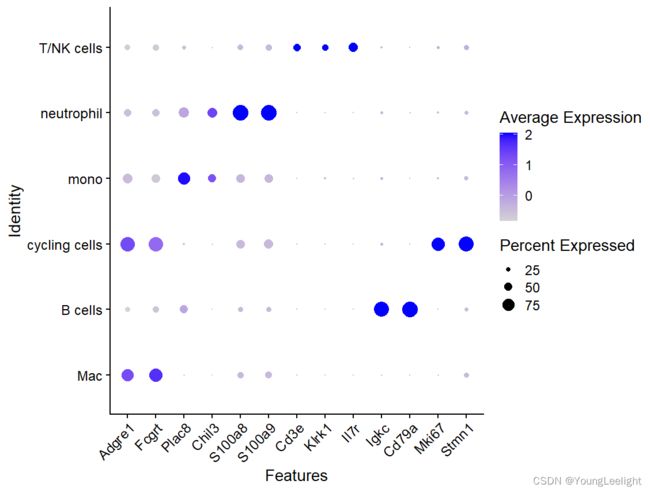
getwd()使用文献里提供的marker gene 进行可视化
MARKER GENE:
macrophages (Adgre1 and Fcgrt), monocytes (Plac8 and Chil3), neutrophils (S100a8/a9), natural killer (NK)/T cells (Cd3e, Klrk1 and Il7r), B cells (Igkc and Cd79a) and cycling cells (Mki67 and Stmn1)
DotPlot(sce.all.int,features = c("Adgre1","Fcgrt",
"Plac8","Chil3",
"S100a8","S100a9",
"Cd3e", "Klrk1","Il7r",
"Igkc","Cd79a",
"Mki67","Stmn1"))

step5: 确定单细胞亚群生物学名字 ######
step5: 确定单细胞亚群生物学名字 ######
# 一般来说,为了节省工作量,我们选择0.1的分辨率进行命名
# 因为命名这个步骤是纯人工 操作
# 除非0.1确实分群太粗狂了,我们就选择0.8
source('scRNA_scripts/lib.R')
# sce.all.int = readRDS('2-harmony/sce.all_int.rds')
colnames([email protected])
DimPlot(sce.all.int,group.by = "orig.ident" )
# 付费环节 800 元人民币
if(F){
sce.all.int
celltype=data.frame(ClusterID=0:9 ,
celltype= 0:9)
celltype
#定义细胞亚群
celltype[celltype$ClusterID %in% c( 5 ),2]='B cells'
celltype[celltype$ClusterID %in% c( 7 ),2]='T/NK cells'
celltype[celltype$ClusterID %in% c( 0,1,4,8 ),2]='Mac'
# celltype[celltype$ClusterID %in% c( 4 ),2]='cycle'
celltype[celltype$ClusterID %in% c( 3 ,9),2]='cycling cells'
celltype[celltype$ClusterID %in% c( 6 ),2]='neutrophil'
#celltype[celltype$ClusterID %in% c( 1 ),2]='endo'
celltype[celltype$ClusterID %in% c( 2 ),2]='mono'
head(celltype)
celltype
table(celltype$celltype)
[email protected]$celltype = "NA"
for(i in 1:nrow(celltype)){
[email protected][which([email protected]$RNA_snn_res.0.1 == celltype$ClusterID[i]),'celltype'] <- celltype$celltype[i]}
Idents(sce.all.int)=sce.all.int$celltype
sel.clust = "celltype"
sce.all.int <- SetIdent(sce.all.int, value = sel.clust)
table([email protected])
dir.create('check-by-celltype')
setwd('check-by-celltype')
source('../scRNA_scripts/check-all-markers.R')
setwd('../')
getwd()
}
###### step6: 单细胞亚群比例差异 ######
# 付费环节 800元人民币
###### step7: 单细胞亚群表达量差异分析 ######
# 付费环节 800 元人民币
其他高级分析
