单细胞组学简介
1. 背景介绍
我们知道生命过程是建立在细胞时空动态调控的基础上的。比如,胚胎发育过程中,随着时间推进,细胞不断分裂、分化、成熟;其过程同时也受到空间信息的调控,比如周边细胞的细胞间通讯。

(Ref: https://en.wikipedia.org/wiki/Human_embryonic_development)
最小的生命单位是细胞,我们可以对细胞进行测序来研究其动态变化过程。05 年左右兴起的 bulk 测序以其高通量的优点迅速成为生命科学领域的重要研究手段,并大大推动了该领域的研究进展。bulk 测序往往是将一群细胞混合在一起测序,难以捕捉细胞间可能存在的异质性。所谓异质性,一般是指不同细胞在某些组学层面(比如基因组、转录组、蛋白质组等)存在显著差异。单细胞测序(比如 09 年开始出现的单细胞转录组测序)可以比较好地解决这个问题。所谓单细胞测序,顾名思义,它可以对单个细胞一一进行测序,从而可以在单细胞精度上研究每个细胞。基于单细胞测序,我们可以利用不同维度的信息(比如不同组学)对细胞进行分群,从而提高研究的“分辨率”。

(Ref: Huang et al, Genome Biology, 2019)
单细胞测序技术发展到今天,多个组学都有了相应的测序平台。
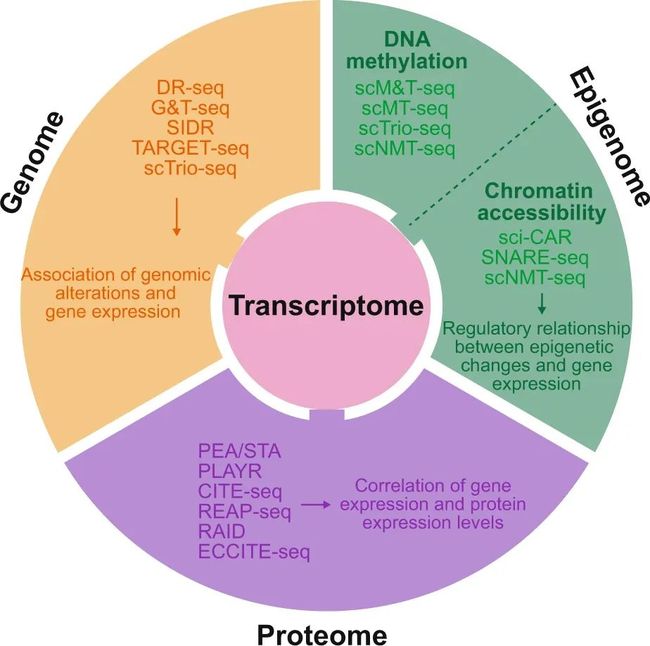
(Ref: Lee et al, Exp Mol Med, 2020)
2. 单细胞转录组测序平台
下文我们主要介绍单细胞转录组测序的基本流程以及应用。
单细胞转录组测序(scRNA-seq)有多个平台,各有特色。根据分离单细胞方法的不同可以分为基于液滴的(droplet-based)以及基于多孔板的(well-based);根据通量大小(一次可以测多少细胞)可以分为高通量平台和低通量平台;根据测序片段覆盖度可以分为全长测序、3’ 端测序或 5’ 端测序。
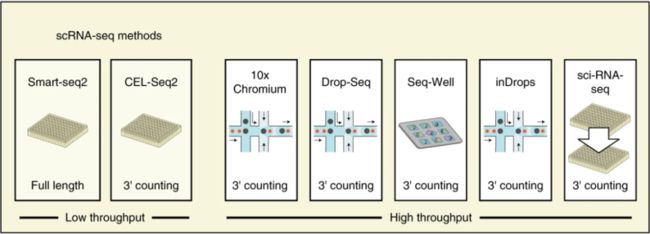
(Ref: Ding et al, Nature Biotechnology, 2020)
以 10x Genomics 平台为例。该平台是目前最流行的 scRNA-seq 平台之一。结合上面的分类方法,它是基于液滴分离单细胞(droplet-based),高通量的,并且是 3’ 端测序或 5’ 端测序(非全长测序)的平台。

(Ref: https://dnacore.missouri.edu/10xgenomics.html)
该平台的大致流程可以概括为:
“将数以百万计的、带有寡核苷酸条形码标签的凝胶珠(Gel Bead)与样品混合 —— 样品可以是高分子量(HMW)DNA、单细胞、经 Feature Barcoding 技术标记的细胞、细胞核、经转座酶处理的细胞核或细胞珠(Cell Beads)。随后将凝胶珠和样品加入油-表面活性剂溶液中,以产生 GEM(Gel Beads in EMulsion),GEM 作为单独的反应囊泡,凝胶珠在其中溶解,样品在其中被加上标签。将带有标签的产物混合,进行下游反应,从而产生与短读长测序仪兼容的文库。测序后,将带有标签的短读长序列交给下游分析流程,软件可利用标签将序列定位到最初的高分子量 DNA、单细胞或单细胞核。”
(Ref: https://pages.10xgenomics.com/rs/446-PBO-704/images/CN_10x_BR025_Chromium-Brochure_A4_Digital.pdf)
3. 单细胞转录组测序的应用
3.1. 细胞类型注释
单细胞测序自问世以来即在生命科学领域得到广泛应用。scRNA-seq 一个最基础的应用就是 cell type annotation:根据其测序结果(transcriptomics profile)对每个细胞进行归类,推断出该细胞属于哪个细胞类型(比如是免疫细胞还是肌细胞)。
scRNA-seq 的输出是一个 cell by gene 的表达量矩阵,其中每个值是 UMI 或者 reads 的数量,用来衡量某个基因在某个细胞中的表达量。为了进行细胞类型注释,基于表达量矩阵,首先从中挑选出高变异基因(highly variable gene)并更新矩阵;然后进行数据降维及聚类;接着找出在不同类中差异表达的基因,如果这些基因恰巧是特定细胞类型的分子 marker,那么可以就此推断这一类细胞属于特定细胞类型。

(Ref: https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html)
如果你对此感兴趣,可以直接查看目前这方面主流分析工具的教程,比如 scanpy 的教程:https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html。
3.2. 肿瘤遗传学
肿瘤研究中一个很重要的主题就是发现亚群并推断其动态变化。一般来说,我们可以通过多个维度的信息(比如体细胞突变:SNP, Indel, CNV 等)对肿瘤细胞进行分群,从而进一步进行肿瘤进化以及耐药机制的研究。

(Ref: Jia et al, Signal Transduction and Targeted Therapy, 2022)
利用 bulk 测序可以推断出一群细胞中不同亚群的比例,但是精度有限,常常只能估算正常细胞与肿瘤细胞的比例而不能对肿瘤细胞进行进一步分群;基于单细胞测序,分群的精度可以大幅提升,从而可能找到一些低丰度的目标亚群(比如下图中基于 scRNA-seq 数据的 CNV 分析找到了两个小亚群,分别只包含 34 个和 7 个细胞)。
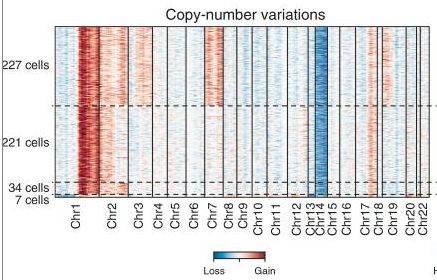
(Ref: Filbin et al, Science, 2018)
3.3.(单时间点)轨迹分析
上文已经提过,生命是一个动态过程。为了研究一个生物过程随时间的动态变化,我们可以在多个时间点进行采样并测序。不过,由于单细胞测序的成本较高或一些技术原因(比如样本珍贵,难以多个时间点采样),通常大家都只会在单个时间点采样测序。一个细胞群体包含了处于多种不同状态(比如不同分化阶段)的细胞,这些细胞在该生物过程中具有不同的变化速度甚至不同的命运。我们可以将在单个时间点测序的结果理解为细胞群体在该时间点的快照(snapshot),这种快照仍然可以捕获特定生物过程中的不同状态。
轨迹推断(trajectory inference)是一种计算方法,通常利用 scRNA-seq 的数据来推断一组细胞在特定生物过程中(例如细胞周期、细胞分化或细胞对外部刺激的反应过程)的隐时间(或者说排序)。它可以被用于谱系发育等研究。
目前,轨迹推断通常基于两种策略,一种是拟时序分析(pseudotime analysis),另一种是 RNA 速度(RNA velocity)分析。
关于 pseudotime analysis,19 年有一篇文章已经对多种相关的分析方法做了详细的比较,这些方法的共同缺点是难以提供可靠的轨迹方向(即难以判别轨迹的哪一端是起点或终点)。

(Ref: Saelens et al, Nature Biotechnology, 2019)
关于 RNA velocity,首先我们回顾一下 RNA 的代谢过程及其动力学:RNA 以速率 合成;然后 RNA 剪接去除内含子并以速率 形成成熟 mRNA;成熟 mRNA 以速率 降解。在稳态下,未剪接和剪接处于平衡状态,即新合成的 RNA 与新剪接的 RNA 数量相等,新剪接的 RNA 与新降解的 RNA 数量相等。因此,未剪接和剪接的 RNA 之间是否处于平衡状态可以作为基因是处于诱导状态还是抑制状态的一个指标。另外,由于速度是矢量,是有方向的,所以 RNA velocity 这种分析方法可以自动判别轨迹的方向。

(Ref: Manno et al, Nature, 2018)
3.4.(多时间点)轨迹分析
相较于单个时间点的测序(snapshot),多个时间点的 scRNA-seq 数据拥有更多信息来进行(发育)轨迹推断。其难点在于如何将多个批次(时间点)的细胞数据联系起来,已有的策略包括:
- 用基于 Optimal Transport 方法找到的不同时间点细胞间的“耦合”关系来拟合真实发育过程中不同时间点细胞的“耦合”关系。
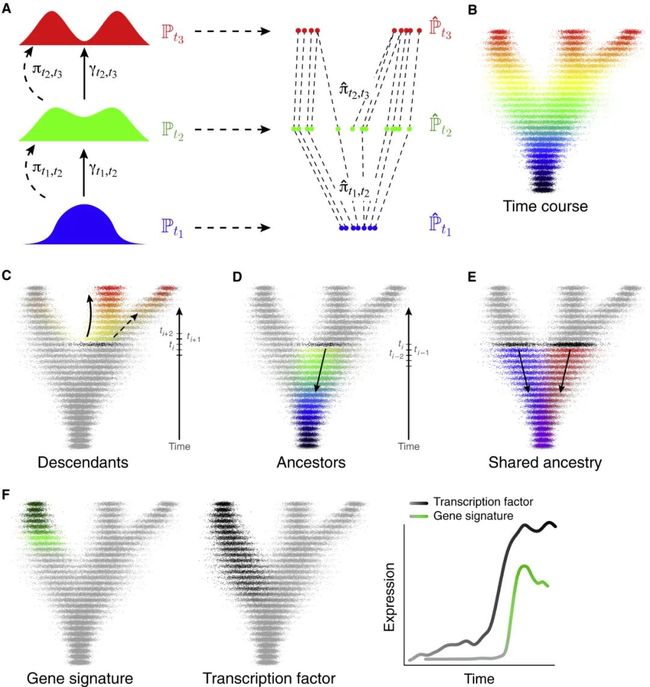
(Ref: Schiebinger et al, Cell, 2019)
- 结合 lineage tracing 的实验技术获得不同时间点细胞间的“耦合”关系。
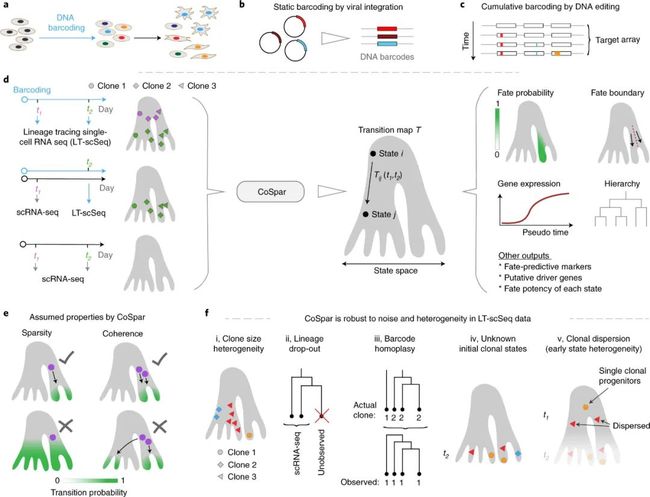
(Ref: Wang et al, Nature Biotechnology, 2022)
4. 单细胞多组学数据整合
最后简单介绍一下单细胞多组学数据的整合。我们知道细胞活动是一个涉及多组学(多层次)的动态过程。在上文第 1 节已经介绍过目前多种组学(比如基因组、转录组、蛋白质组等)都有相应的单细胞测序平台,那么将这些多组学的数据整合在一起往往会描绘出一幅更完整的细胞(多层次)变化的图谱。
下图是一个很好的示意图,其中每一行是一批细胞,每一列是一种组学的特征(feature);不同批次的细胞数可能不一样(即 ,, 可能不相等);不同组学的特征数量也可能不相等(比如 scRNA-seq 中的 gene 数量很可能不等于 scATAC-seq 中的 peak 数量)。如果横向整合,代表整合同一批次细胞不同组学的数据;MIRA(Lynch et al, 2022)就是解决这一类任务的一种工具。如果纵向整合,代表整合不同批次细胞的同一种组学数据;某种程度上我们联想到 3.4 小节中的多时间点 scRNA-seq 数据的分析。还有一种情形是对角线整合,也就是整合不同批次的不同组学的数据,这一类任务最具挑战性,不过已经有相应的分析工具了,比如 GLUE(Cao and Gao, 2022)。
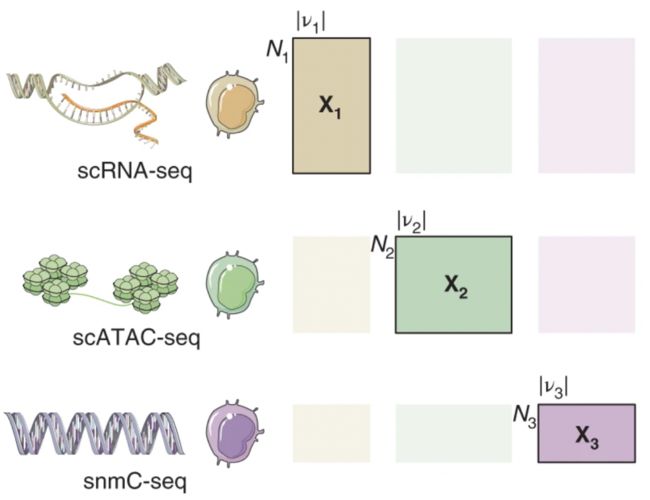
(Ref: Cao and Gao, Nature Biotechnology, 2022)
5. 小结
本文简单介绍了单细胞测序的技术背景、原理和应用。从 bulk 测序到单细胞测序,研究的“分辨率”大大提升;单细胞测序在生命科学多个领域得到了广泛应用。生命是一个在时空维度上动态变化的过程。单细胞测序是探索细胞在时间维度上动态变化的强大工具,不过受限于其技术原理,它难以对细胞在空间维度上的动态变化进行研究。空间转录组(spatial transcriptomics)技术是一种可以保留细胞空间信息的”单细胞“测序平台,近几年已经引起越来越多的关注,可以预期未来几年整合 scRNA-seq 与 spatial transcriptomics 的数据并据此研究细胞的时空动态变化会是一个热点。
参考资料
- [1] https://en.wikipedia.org/wiki/Human_embryonic_development
- [2] Huang et al, Genome Biology, 2019. https://doi.org/10.1186/s13059-019-1865-2
- [3] Lee et al, Exp Mol Med, 2020. https://doi.org/10.1038/s12276-020-0420-2
- [4] Ding et al, Nature Biotechnology, 2020. https://doi.org/10.1038/s41587-020-0465-8
- [5] https://dnacore.missouri.edu/10xgenomics.html
- [6] https://pages.10xgenomics.com/rs/446-PBO-704/images/CN_10x_BR025_Chromium-Brochure_A4_Digital.pdf
- [7] https://scanpy-tutorials.readthedocs.io/en/latest/pbmc3k.html
- [8] Jia et al, Signal Transduction and Targeted Therapy, 2022. https://doi.org/10.1038/s41392-022-00990-4
- [9] Filbin et al, Science, 2018. https://www.science.org/doi/10.1126/science.aao4750
- [10] Saelens et al, Nature Biotechnology, 2019. https://doi.org/10.1038/s41587-019-0071-9
- [11] Manno et al, Nature, 2018. https://doi.org/10.1038/s41586-018-0414-6
- [12] Schiebinger et al, Cell, 2019. https://doi.org/10.1016/j.cell.2019.01.006
- [13] Wang et al, Nature Biotechnology, 2022. https://doi.org/10.1038/s41587-022-01209-1
- [14] Lynch et al, Nature Methods, 2022. https://doi.org/10.1038/s41592-022-01595-z
- [15] Cao and Gao, Nature Biotechnology, 2022. https://doi.org/10.1038/s41587-022-01284-4