宏病毒组介绍
病毒是一种个体微小,结构简单,只含一种核酸(DNA 或 RNA),必须在活细胞内寄生并以复制方式增殖的非细胞型生物。病毒是地球上丰度最高的“生物”类群,它们广泛地存在于所有已知的生态环境中,从水体,土壤再到人体,病毒无处不在。病毒能够感染所有已知的生物类型,从动物、植物到微生物,包括细菌和古菌。
人体内的病毒主要包括真核病毒和噬菌体,按核酸类型,可分为双链DNA(double-stranded DNA,dsDNA)病毒,单链 DNA(single-stranded DNA ,ssDNA)病毒和RNA 病毒。
随着病毒研究的深入以及宏基因组学的快速发展,“宏病毒组”和“宏病毒组学”的概念应用而生。
宏病毒组学(Viral Metagenomics)是在宏基因组学理论的基础上,结合现有的病毒分子生物学检测技术而兴起的一个新的学科分支。 宏病毒组直接以样本中所有病毒的遗传物质为研究对象,能够快速准确的鉴定样本中所有的病毒组成——也常被称为“病毒组(Virome)”。宏病毒组学应用广泛,可应用于人或动物肠道或者血液样本、水样、土壤样本等的研究。不仅在病毒发现、病毒溯源、微生物预警等研究方面具有重要作用,在病毒的起源和进化模式、遗传多样性和地理分布等研究方面也具有重要意义。
宏病毒组研究始于 2002 年,Bretbart 等人利用宏基因组学技术研究了两个未培养的海洋病毒群落,发现噬菌体为海水中主要病毒类型。近年来,宏病毒组研究呈爆炸式增长,人们越来越意识到病毒在调节微生态平衡上发挥着重要作用。
但是由于受到传统病毒培养方法的限制,目前已知的病毒种类只有6000多种,而高通量测序技术的发展和广泛应用,越来越多的未知病毒被发现。定于2018年启动的“全球病毒组项目(Global Virome Project)”计划耗资12亿美元,旨在鉴定仍未“现身”的约百万种病毒(其中40%—50%的病毒可能感染人类),将宏病毒组研究推到了新的热潮。
在宏基因组数据中,宿主和细菌序列的占比高达99%,病毒只占到万分之几的水平。而直接提取环境或组织中宏病毒DNA或RNA序列的研究较少,且提取成功率也很低(通常不到1%)。
微基生物的宏病毒组技术路线如下
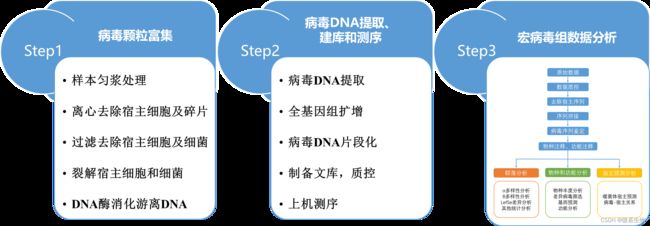

简言之,就是使用特殊方法把特定样本中所有病毒与其他微生物分开,然后提取病毒DNA,建库测序后进行病毒序列鉴定,之后对鉴定的病毒序列进行 vOTUs 分析,并对得到的 vOTUs 序列进行物种和功能上的注释以及分类,包括 NR,COG,KEGG,pfam 等。在上述分析的基础上进行病毒新颖性分析,进化树构建,宿主预测以及病毒与群落中细菌、古菌等其他微生物之间的互作等分析。
微基生物样本病毒颗粒富集主要包括五个关键步骤:
1. 样本匀浆处理,旨在使病毒颗粒尽可能多地释放出来;
2. 离心去除宿主细胞和碎片;
3. 0.22μm过滤去除细菌的干扰;
4. 进一步裂解过滤液中的宿主细胞和细菌;
5. 使用核酸酶消化游离的DNA和RNA,去除核酸污染。
病毒序列鉴定是宏病毒组生信分析中的关键一环,微基生物使用多种软件对组装序列进行病毒鉴定,并对不同软件鉴定出来的病毒序列作出统计。
病毒鉴定软件 1 :VirFinder 工具
病毒鉴定软件 2 :VirSorter2 工具
病毒鉴定软件 3 :CAT 工具
病毒鉴定软件 4 :IMG/VR 数据库比对
文献解读1:Intestinal Virome in Patients With Alcoholic Hepatitis
酒精性肝炎患者的肠道病毒组
Hepatology IF:17.4251
酒精性肝炎(Alcoholic hepatitis,AH)是酒精相关性肝病(Alcohol-associated liver disease,ALD)的一种严重表现,死亡率高。虽然肠道细菌和真菌可以影响疾病的发生,但对ALD患者病毒组的影响知之甚少。为此来自加州大学圣地亚哥的学者通过分析AH患者及对照组的粪便病毒组,探究酒精性肝炎患者的肠道病毒组特征。
该研究共纳入89例AH患者,36例酒精不耐受(Alcohol use disorder,AUD)患者和17例健康对照组,从粪便样本中分离出病毒样颗粒,通过宏基因组测序对肠道病毒组进行分析。
研究表明,相较于对照组,酒精性肝病患者的粪便病毒多样性显著升高,埃希氏杆菌属、肠杆菌属及肠球菌属噬菌体显著增加,细小DNA病毒科及疱疹病毒科也显著增加;在AH患者的粪便样本中,病毒多样性变化最显著,大肠杆菌、肠杆菌和肠球菌噬菌体的数量过多,细菌多样性降低。抗生素治疗与更高的粪便病毒多样性相关;葡萄球菌属与疱疹病毒科等特定病毒分类群与酒精性肝病严重程度及90天死亡率的增加相关。
图1. 酒精性肝炎的严重程度与粪便病毒组的改变相关(如下图)
(a) 在科水平上,三组粪便样本中肠道噬菌体、哺乳动物病毒和其他病毒的相对丰度;(b) 基于香农指数和逆辛普森指数的病毒多样性,以及基于 Chao1 指数的病毒丰富度。(c) 根据细菌宿主的不同,在物种水平上,三组中噬菌体的平均相对丰度,0–1 对应于 0–100% 丰度;(d)线性判别效应大小分析 (LEfSe) 识别最有可能解释三组之间差异的特征:在科、属和种水平上噬菌体的相对丰度以及在科和属水平上的哺乳动物病毒;(e)在科水平上,每组中哺乳动物病毒呈阳性的受试者百分比; (f) 87 名酒精性肝炎患者粪便样本疱疹病毒科阳性或阴性的 MELD 评分。
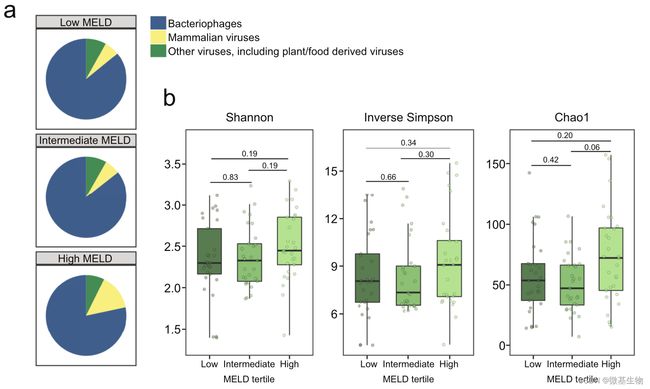
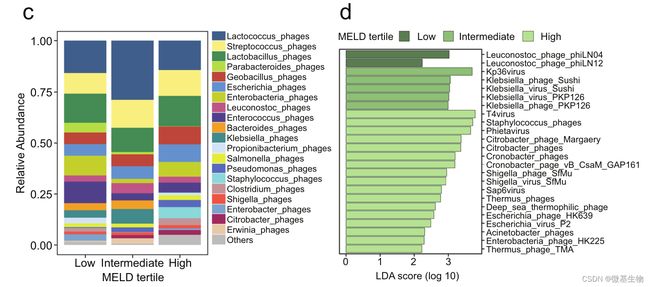

图1. 酒精性肝炎的严重程度与粪便病毒组的改变相关
文献解读2
Viromes outperform total metagenomes in revealing the spatiotemporal patterns of agricultural soil viral communities
在农业土壤病毒组时空模式研究中病毒组优于宏基因组
ISME IF:10.302
摘要:
土壤中的病毒含量丰富且在生物地球化学循环中扮演重要角色,但目前对病毒的研究还不够深入。本研究在生物炭改良后的土壤中种植番茄,并在种植前和收获时收集土壤样本同时进行宏基因组和宏病毒组测序以研究土壤样本中的病毒群落。研究结果表明,对土壤样本进行病毒富集后提取DNA的宏病毒组方法能有效减少病毒组中非病毒序列,大大提高病毒序列的比例,获得更全面的病毒信息(vOTU)和更准确的病毒多样性结果。在检测到的2961个vOTU中有2684个vOTU是只来自于宏病毒组的分析结果,仅有3个vOTU是只来自于宏基因组的分析结果。研究结果突出了宏病毒组在研究土壤中病毒的有效应用,并揭示了在番茄整个生长周期中土壤病毒和微生物群落之间的动态相关性。
病毒富集方法:
本研究在番茄种植前(4月)和收获时(8月)分别在不同位置收集了16个土壤样本,分别取部分进行常规的宏基因组实验和检测,同时分别取适量样本进行病毒富集,病毒富集方法如下:
首先重悬土壤样本,使用0.22m滤膜过滤,其次使用超速离心法浓缩病毒颗粒,然后用核酸酶去除游离DNA,最后使用Powersoil试剂盒提取病毒DNA。
主要结果
1. 宏病毒组研究土壤病毒群落更有优势
在本研究中,4月与8月样本的宏基因组下机数据分别为8741015 reads和14551631 reads,而4月与8月样本的宏病毒组的下机数据为9519518 reads和5770419 reads。宏病毒组组装得到总长度为800 Mb的169421条contig,而宏基因组仅组装得到总长度为65 Mb的22951条contig。基于以上组装结果分别基于DeepVirFinder和VirSorter进行病毒鉴定,发现宏病毒组中52.4%的contigs被鉴定为病毒,而宏基因组仅有2.2%(图1)。可见前期的病毒富集处理可大幅度提高病毒的有效数据量,同时有助于提高组装的质量。
对聚类得到的共105909条contig按照vOTU的思路进行鉴定,共得到2961个vOTU,其中2864个vOTU仅在宏病毒组中存在,94个vOTU同时存在于宏病毒组和宏基因组,而仅有3个vOTU只存在宏基因组。对上述的2961个vOTU进行丰度分析,发现同时存在于宏病毒组和宏基因组的94个vOTU的丰度普遍较高,表明从宏基因组中难以获知低丰度病毒的信息。
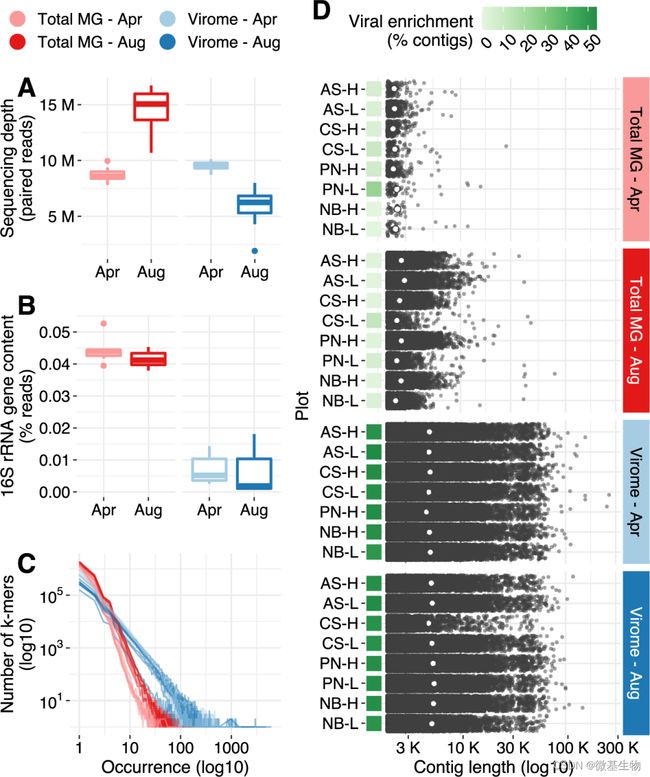 图1. 宏病毒组与宏基因组的病毒鉴定情况
图1. 宏病毒组与宏基因组的病毒鉴定情况
A. 跨分析方法和时间点的测序深度分布。 B. 分类为 16S rRNA 基因片段的读数百分比。C. 序列复杂性,通过在每个库中检测到的一组代表性 k-mers (k = 31) 的频率分布来衡量。D 组装的重叠群的长度分布(最小长度 = 2Kbp)。
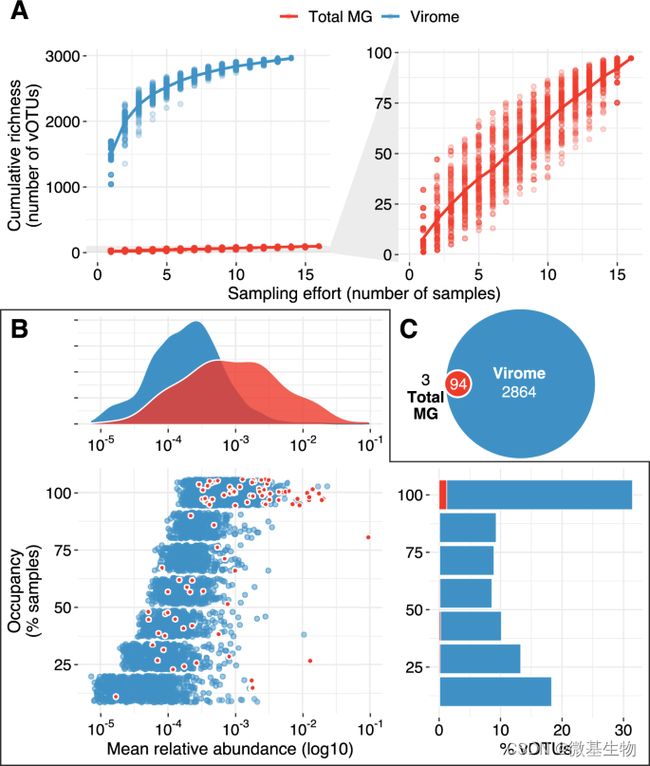 图2. 宏病毒组和宏基因组方法获得的病毒丰富度。
图2. 宏病毒组和宏基因组方法获得的病毒丰富度。
A. 宏基因组(红色,n = 16)和宏病毒组(蓝色,n = 14)中 vOTU 的累积曲线.
B. 宏病毒组的 vOTU 的丰度。蓝色的是仅在宏病毒组中检测到的 vOTU,红色的是在宏病毒组和宏基因组中同时检测到的 vOTU。
C. vOTU (n = 2961) 的检测重叠。红色表示两种分析方法均检测到的 vOTU,有三个vOTU仅在宏基因组中检测。
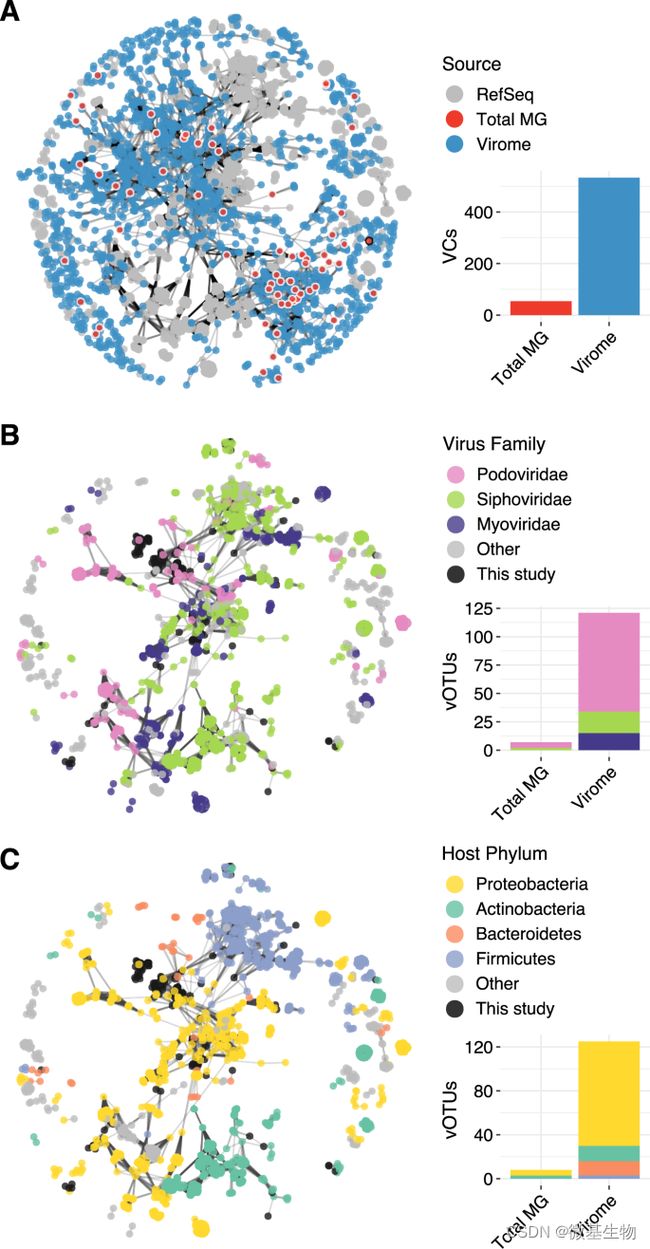
图3.宏病毒组和宏基因组鉴定的病毒多样性和预测的病毒(vOTU) 宿主。
2. 病毒与微生物在时间与空间中的动态关系
病毒依赖于微生物宿主进行复制,因此在4月和8月的样本的病毒组成和微生物组成中观察到了明显的相关性。同时对土壤环境因子与病毒和微生物组成进行相关性分析,发现存在明显的时间梯度的显著关联,如4月样本中环境因子与群落的变化规律与8月样本中观察到的完全相反。
本研究中的土壤样本存在东西方向与南北方向的位置特征(图4B),使用各样本的vOTU数据进行基于Bray-Curtis的主成分分析(图4A)发现在PCo1轴观察到明显的不同时间点样本的分离情况,PCo2轴却观察到明显的东西位置点样本的差异。深入分析发现共有1035个vOTU的相对丰度出现东西方向梯度的变化,其中460个vOTU的丰度从东到西呈现递增的趋势,而575个vOTU的丰度则出现了从西到东的递增趋势(图4D),结合样本的收集时间点,发现分别在4月和8月样本中存在从东到西递增规律的vOTU之间基本不重叠。但对微生物群落分析却发现不存在如病毒群落相似的东西方向的丰度变化趋势,推测可能是土壤中在实验前残留的病毒的衰变周期较长所导致,同时亦可能受到东西方向的边界环境影响,如道路扬尘等因素。
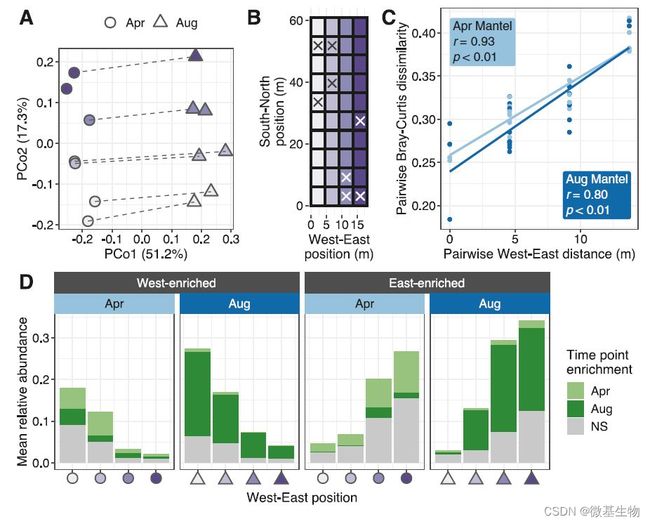
图4. 病毒群落空间结构
结论:宏病毒组的病毒富集前处理可有效提高有效的病毒数据比例,在分析结果中可更全面的还原样本中的病毒组成和丰度情况。病毒与微生物群落之间存在密切的相关性,但病毒群落同时亦会具有自身的固有特性,并且易受外界条件影响。