理解:RPM、RPKM、FPKM、TPM、DESeq、TMM、SCnorm、GeTMM、ComBat-Seq
文章目录
-
- 前言
- 为什么有不同的标准化表达单位(Expression Units)
- 理解Expression Units 和计算Calculation
-
- RPM or CPM
-
- 例子:RPM or CPM normalization 使用Python Tool:[`bioinfokit`](https://github.com/reneshbedre/bioinfokit)
- RPKM (Reads per kilo base per million mapped reads)
-
- 例子RPKM
- 例子:RPKM or FPKM normalization 使用 Python Tool:[`bioinfokit`](https://github.com/reneshbedre/bioinfokit)
- TPM (Transcripts per million)
-
- 例子:TPM normalization 使用Python Tool:[`bioinfokit`](https://github.com/reneshbedre/bioinfokit)
- 手动计算比较RPKM和TPM(python)
- TMM (Trimmed Mean of M-values)
-
- TMM计算:
- TMM 的实现:`edgeR`
- DESeq or DESeq2 标准化 (median-of-ratios method)
-
- DESeq2 normalization :使用R Package :`DESeq2`
- SCnorm for single cell RNA-seq (scRNA-seq)
- ComBat-Seq method
- GeTMM method
-
- GeTMM normalization :R package `edgeR`
- 参考

前言
在RNAseq 等二代测序数据的分析中,总会遇到各种各样的基因表达单位:RPM, RPKM, FPKM, TPM, TMM, DESeq, SCnorm, GeTMM, ComBat-Seq 和 Raw Reads Counts等(就像长度单位厘米)。Expression units 提供了基因或转录本等相对丰度的数学度量。大多数时候很难理解这些Expression units怎么从Raw count算来的。
在这里进行简单的总结整理。欢迎批评指正
公众号:猪猪的乌托邦
为什么有不同的标准化表达单位(Expression Units)
- 对原始数据的标准化是必要的,测序深度越深,在同一水平上表达的基因reads 就会越多,基因越长,相同水平的reads 会越多。标准化的Expression Units 可以一定程度上消除实验技造成的偏差,如测序深度、基因长度等,并使数据在样本内和样本间具备直接的可比性。(expression units,可以说是标准化的表现形式)
- Normalized expression units 有助于消除批次效应,使数据具备可比性。
理解Expression Units 和计算Calculation
RPM or CPM
RPM:Reads per million mapped reads ;CPM: Counts per million mapped reads
R P M o r C P M = R e a d s N u m b e r o f a G e n e × 1 0 6 T o t a l n u m b e r o f M a p p e d R e a d s RPM or CPM = \frac{Reads\,Number\,of\,a\,Gene\times 10^6}{Total\,number\,of\,Mapped\, Reads } RPMorCPM=TotalnumberofMappedReadsReadsNumberofaGene×106
比如: 测 5 Million(M) reads 的库,其中 4 M 比对到基因组上并且5000 Reads 匹配到某基因。此时RPM or CPM: 5000 × 1 0 6 4 × 1 0 6 = 1250 \frac{5000 \times 10^6} {4 \times 10^6} = 1250 4×1065000×106=1250
Tips:
- 不考虑对基因或转录长度的标准化
- 适合未考虑基因长度的方法获取的的Raw Reads Counts
例子:RPM or CPM normalization 使用Python Tool:bioinfokit
from bioinfokit.analys import norm, get_data
# 载入甘蔗的RNAseq原始基因表达矩阵(Bedre et al.,2019)
df = get_data('sc_exp').data
df.head(2)
# 输出
gene ctr1 ctr2 ctr3 trt1 trt2 trt3 length
0 Sobic.001G000200 338 324 246 291 202 168 1982.0
1 Sobic.001G000400 49 21 53 16 16 11 4769.0
# 矩阵中有长度这一列,先删掉
df = df.drop(['length'], axis=1)
# 将gene列作为索引列
df = df.set_index('gene')
df.head(2)
# 输出
ctr1 ctr2 ctr3 trt1 trt2 trt3
gene
Sobic.001G000200 338 324 246 291 202 168
Sobic.001G000400 49 21 53 16 16 11
# now, normalize raw counts using CPM method
nm = norm()
nm.cpm(df=df)
# get CPM normalized dataframe
cpm_df = nm.cpm_norm
cpm_df.head(2)
# 输出
ctr1 ctr2 ctr3 trt1 trt2 trt3
gene
Sobic.001G000200 100.695004 101.731189 74.721094 92.633828 74.270713 95.314714
Sobic.001G000400 14.597796 6.593688 16.098447 5.093269 5.882829 6.240844
RPKM (Reads per kilo base per million mapped reads)
R P K M = R e a d s N u m b e r o f g e n e × 1 0 3 × 1 0 6 T o t a l n u m b e r o f M a p p e d R e a d s × g e n e l e n g t h i n b p RPKM = \frac{Reads\,Number\,of gene \times10^3\times10^6}{Total\,number\,of\,Mapped\, Reads \times gene\,length\,in\,bp} RPKM=TotalnumberofMappedReads×genelengthinbpReadsNumberofgene×103×106
1 0 3 10^3 103对基因长度normalize, 1 0 6 10^6 106针对测序深度的因素,RPKM 单端
FPKM (Fragments per kilo base per million mapped reads) 与RPKM 类似,针对双端测序数据,在双端测序中,(左右)两条Reads来自同一个DNA Fragment,双端数据做比对时,一个Fragment中的两条Reads 或者只有一个高质量的Reads可以比对到参考基因组上。为了避免混淆和重复计数,对两个或单个Reads比对上的Fragment进行Count,并用于FPKM计算。
例子RPKM
测了一个5 Million(M) Reads 的库,其中,共 4 M 比对到基因组上,并且5000 Reads 匹配到某基因,该基因长度为2000bp。那么,RPKM的计算方式为: 5000 × 1 0 3 × 1 0 6 4 × 1 0 6 × 2000 = 625 \frac{5000 \times10^3\times 10^6} {4 \times 10^6\times2000} = 625 4×106×20005000×103×106=625
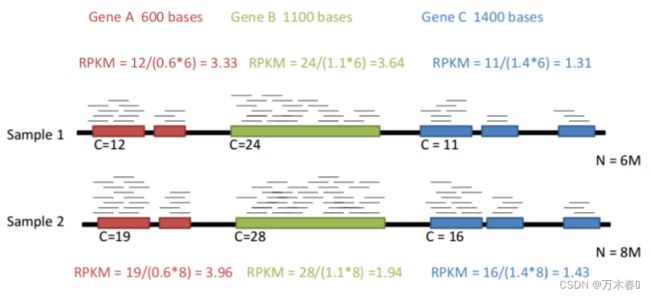
Notes:
- RPKM Normalize的时候考虑了基因长度
- RPKM 适合有基因长度的测序数据
- RPKM单端测序 ,FPKM双端测序
Tips: RPKM/FPKM不代表相对RNA摩尔浓度(rmc)的真值,由于每个样本的总标准化计数将不同,此方法倾向应用于差异表达基因的鉴定。建议使用TPM作为RPKM的替代方法。
例子:RPKM or FPKM normalization 使用 Python Tool:bioinfokit
# 还是前面的数据
df.head(2)
# output
ctr1 ctr2 ctr3 trt1 trt2 trt3 length
gene
Sobic.001G000200 338 324 246 291 202 168 1982.0
Sobic.001G000400 49 21 53 16 16 11 4769.0
# now, normalize raw counts using RPKM method
# gene的长度单位必须是bp
nm = norm()
nm.rpkm(df=df, gl='length')
# get RPKM normalized dataframe
rpkm_df = nm.rpkm_norm
rpkm_df.head(2)
# output
ctr1 ctr2 ctr3 trt1 trt2 trt3
gene
Sobic.001G000200 50.804745 51.327542 37.699846 46.737552 37.472610 48.090169
Sobic.001G000400 3.060976 1.382614 3.375644 1.067995 1.233556 1.308627
PS:…有人认为RPKM/FPKM 没有意义,不是很合理,简书上黄树嘉 的文章做了总结……
TPM (Transcripts per million)
T P M = A ∑ A × 1 0 6 TPM = \frac{A}{\sum A} \times 10^6 TPM=∑AA×106
W h e r e A = R e a d s N u m b e r o f g e n e × 1 0 3 g e n e l e n g t h i n b p Where A = \frac{Reads\,Number\,of\,gene\times10^3}{gene\,length\,in\,bp} WhereA=genelengthinbpReadsNumberofgene×103
TPM与RPKM 转换
T P M = R P K M ∑ R P K M × 1 0 6 TPM = \frac{RPKM}{\sum RPKM} \times10^6 TPM=∑RPKMRPKM×106
Notes:
- TPM normalization 考虑了基因长度
- TPM 适合有基因长度的测序数据
相对于RPKM度量的不精确性,建议将TPM作为RPKM的替代方法。与RPKM相比,TPM平均值是恒定的,并且与相对RNA摩尔浓度(rmc)成比例
例子:TPM normalization 使用Python Tool:bioinfokit
# 接前面
nm.tpm(df=df, gl='length')
# get TPM normalized dataframe
tpm_df = nm.tpm_norm
tpm_df.head(2)
# output
ctr1 ctr2 ctr3 trt1 trt2 trt3
gene
Sobic.001G000200 99.730156 97.641941 72.361658 89.606265 69.447237 90.643338
Sobic.001G000400 6.008723 2.630189 6.479263 2.047584 2.286125 2.466582
手动计算比较RPKM和TPM(python)
import pandas as pd
import numpy as np
def read_counts2tpm(df, sample_name):
"""
convert read counts to TPM (transcripts per million)
:param df: a dataFrame contains the result coming from featureCounts
:param sample_name: a list, all sample names, same as the result of featureCounts
:return: TPM
"""
result = df
sample_reads = result.loc[:, sample_name].copy()
gene_len = result.loc[:, ['Length']]
rate = sample_reads.values / gene_len.values
tpm = rate / np.sum(rate, axis=0).reshape(1, -1) * 1e6
return pd.DataFrame(data=tpm, columns=sample_name, index=df['Gene'])
def read_counts2rpkm(df, sample_name):
result = df
sample_reads = result.loc[:, sample_name].copy()
gene_len = result.loc[:, ['Length']]
total_reads = np.sum(sample_reads.values, axis=0).reshape(1, -1)
rate = sample_reads.values / gene_len.values
tpm = rate / total_reads * 1e6
return pd.DataFrame(data=tpm, columns=sample_name, index=df['Gene'])
# raw data
a = pd.DataFrame(data = {
'Gene': ("A","B","C","D","E"),
'Length': (100, 50, 25, 5, 1),
'S1': (80, 10, 6, 3, 1),
'S2': (20, 20, 10, 50, 400)
})
tpm = read_counts2tpm(a, ['S1', 'S2'])
rpkm = read_counts2rpkm(df=a, sample_name=['S1', 'S2'])
# TPM
Gene S1 S2
A 281690.140845 486.618005
B 70422.535211 973.236010
C 84507.042254 973.236010
D 211267.605634 24330.900243
E 352112.676056 973236.009732
# RPKM
Gene S1 S2
A 8000.0 400.0
B 2000.0 800.0
C 2400.0 800.0
D 6000.0 20000.0
E 10000.0 800000.0
b = pd.DataFrame(data = {
'RPKM_col_sum': (rpkm['S1'].sum(),
rpkm['S2'].sum()),
'TPM_col_sum': (tpm['S1'].sum(),
tpm['S2'].sum())
})
b
# 比较就可以发现TPM的优势,每个样本总的TPM值是相同的,这样的结果便于样本间差异的比较
RPKM_col_sum TPM_col_sum
0 28400.0 1000000.0
1 822000.0 1000000.0
TMM (Trimmed Mean of M-values)
- 区别去之前提到的样本内标准化方法,TMM 是一种样本间的标准化方法
- TMM 假定大部分基因的表达在样本间是没有差异的
- TMM 对样本间的总Reads Counts进行normalize, 而且不对基因长度或者library的大小进行normalize处理
- TMM considers sample RNA population and effective in normalization of samples with diverse RNA repertoires (e.g. 不同组织样本). 在比较不同组织或基因型的样品,或者在样本间RNA Population 有显著差异时,TMM在处理数据批次效应上,具备一定的优势。
- TMM在样本间比较方面表现更好,可以使用R package:
edgeR实现, edgeR假定基因长度在样本之间是恒定的,不考虑对基因长度normalize
TMM计算:
- 获取每个样本中每个基因的library size normalized read count
- 计算两个样本间的Log 2 _2 2 Fold Change (M value) :
M = log 2 T r e a t e d S a m p l e C o u n t s C o n t r o l S a m p l e C o u n t s M = \log_2\frac{Treated\,Sample\,Counts}{Control\,Sample\,Counts} M=log2ControlSampleCountsTreatedSampleCounts - 获取绝对表达量 (A value):
A = log 2 ( T r e a t e d S a m p l e C o u n t s ) + log 2 ( C o n t r o l S a m p l e C o u n t s ) 2 A= \frac{\log_2(Treated\,Sample\,Counts) + \log_2(Control\,Sample\,Counts)}{2} A=2log2(TreatedSampleCounts)+log2(ControlSampleCounts) - 处理数据(去除M值的±30%,去除A值的±5%)
- 修剪后得到M的加权平均值,计算Normalize Factor (Robinson et al., 2010 for details)
TMM 的实现:edgeR
# load library
library(edgeR)
x <- read.csv("https://reneshbedre.github.io/assets/posts/gexp/df_sc.csv",row.names="gene")
# delete last column (gene length column)
x <- x[,-7]
head(x)
ctr1 ctr2 ctr3 trt1 trt2 trt3
Sobic.001G000200 338 324 246 291 202 168
Sobic.001G000400 49 21 53 16 16 11
Sobic.001G000700 39 49 30 46 52 25
Sobic.001G000800 530 530 499 499 386 264
Sobic.001G001000 12 3 4 3 10 7
Sobic.001G001132 4 2 2 3 4 1
group <- factor(c('c','c', 'c', 't', 't', 't'))
y <- DGEList(counts=x, group=group)
# normalize for library size by cacluating scaling factor using TMM (default method)
y <- calcNormFactors(y)
# normalization factors for each library
y$samples
group lib.size norm.factors
ctr1 c 3357347 1.0290290
ctr2 c 3185467 0.9918449
ctr3 c 3292872 1.0479952
trt1 t 3141934 0.9651681
trt2 t 2720231 0.9819187
trt3 t 1762881 0.9864858
# count per million read (normalized count)
norm_counts <- cpm(y)
head(norm_counts)
ctr1 ctr2 ctr3 trt1 trt2 trt3
Sobic.001G000200 97.860339 102.5561297 71.3023988 95.9799323 75.634827 96.6223700
Sobic.001G000400 14.186854 6.6471566 15.3618989 5.2772471 5.990877 6.3264647
Sobic.001G000700 11.291578 15.5100320 8.6954145 15.1720855 19.470352 14.3783289
Sobic.001G000800 153.449643 167.7615701 144.6337277 164.5841451 144.529917 151.8351528
Sobic.001G001000 3.474332 0.9495938 1.1593886 0.9894838 3.744298 4.0259321
Sobic.001G001132 1.158111 0.6330625 0.5796943 0.9894838 1.497719 0.5751332
DESeq or DESeq2 标准化 (median-of-ratios method)
- DESeq (DESeq2) 标准化方法和TMM类似
- DESeq normalization 方法同样假定大部分基因的表达是不存在差异的
- The DESeq 计算每个样本的大小因子,比较不同样本在不同不同深度下的Counts
- DESeq normalization uses the median of the ratios of observed counts to calculate size factors.
通过将每个样品的Counts除以其几何平均值来计算比值
然后将 Size Factor 定义为每个样本的该比值的中位数。
接下来使用Size Factor ** 对每个样本的原始计数矩阵(Raw Count Data) **执行标准化 - 同样,DESeq or DESeq2 不考虑基因长度的影响,并假定基因长度在每个样本之间都是恒定的
- DESeq or DESeq2 更适用于样本间比较
Note: 使用 DESeq2 进行分析的时候,需要整数形式的数据作为输入。如果用RSEM(RNA-Seq by Expectation-Maximization),建议使用
tximport导入数据,再使用DESeq2的DESeqDataSetFromTximport()执行接下来的基因表达水平分析。此外,也可以使用 RSEM 的近似整数矩阵,但会失去tximport提供的例如对每个基因转录长度的Normalize。
DESeq2 normalization :使用R Package :DESeq2
library(DESeq2)
x <-read.csv("https://reneshbedre.github.io/assets/posts/gexp/df_sc.csv",row.names="gene")
cond <- read.csv("https://reneshbedre.github.io/assets/posts/gexp/condition.csv",row.names="sample")
cond$condition <- factor(cond$condition)
x <- x[, rownames(cond)]
head(x)
ctr1 ctr2 ctr3 trt1 trt2 trt3
Sobic.001G000200 338 324 246 291 202 168
Sobic.001G000400 49 21 53 16 16 11
Sobic.001G000700 39 49 30 46 52 25
Sobic.001G000800 530 530 499 499 386 264
Sobic.001G001000 12 3 4 3 10 7
Sobic.001G001132 4 2 2 3 4 1
# get dds
dds <- DESeqDataSetFromMatrix(countData = x, colData = cond, design = ~ condition)
dds <- estimateSizeFactors(dds)
# DESeq2 normalization counts
y <- counts(dds, normalized = TRUE)
head(y)
ctr1 ctr2 ctr3 trt1 trt2
Sobic.001G000200 272.483741 290.412982 199.133348 272.915069 211.917896
Sobic.001G000400 39.502081 18.823064 42.902713 15.005640 16.785576
Sobic.001G000700 31.440432 43.920482 24.284555 43.141214 54.553122
Sobic.001G000800 427.267404 475.058273 403.933092 467.988384 404.952020
Sobic.001G001000 9.673979 2.689009 3.237941 2.813557 10.490985
Sobic.001G001132 3.224660 1.792673 1.618970 2.813557 4.196394
trt3
Sobic.001G000200 271.037655
Sobic.001G000400 17.746513
Sobic.001G000700 40.332984
Sobic.001G000800 425.916314
Sobic.001G001000 11.293236
Sobic.001G001132 1.613319
# get size factors
sizeFactors(dds)
ctr1 ctr2 ctr3 trt1 trt2 trt3
1.2404410 1.1156526 1.2353531 1.0662658 0.9531993 0.6198401
SCnorm for single cell RNA-seq (scRNA-seq)
- 以上都是针对bulk RNA-seq ,但是应用于 scRNA-seq(单细胞的)就会存在偏差。因为单细胞数据存在大量的非零Counts、Counts与测序深度的关系多变性(基因表达对测序深度的依赖性)、以及其他的技术因素
- Bacher et al., 2017 ,提出了SCnorm—— 一个针对scRNA-seq数据的稳定而准确的样本间 标准化单位(Normalization Unit)
- SCnorm normalization大致步骤
- 使用**
RSEM****featureCounts****Rsubread****或者 **HTSeq等获取表达矩阵,取其近似估计值 - 过滤掉低表达的基因 (Counts至少为10)
- 使用分位数回归估计 Counts和测序深度的关系
- 使用Count-Depth关系,对基因的表达进行聚类
- 针对每个类群计算缩放比例(scale factor ),用于对表达矩阵的normalize
- 使用**
- SCnorm可以在 Bioconductor 中获取
ComBat-Seq method
- Zhang et al., 2020 提出ComBat-Seq (批次效应矫正方法) 以解决RNA-seq测序中由于批次效应引起的差异
- ComBat-Seq 的优势在于矫正了原始数据(Raw Counts)中的批次效应,比如技术性差别:不同的测序仪器或者不同的公司的试剂、不同的实验人员等,并且ComBat-Seq与上述方法都不同(e.g. RPKM, TPM, TMM),其提供了整数形式的数据矩阵,校正后的数据可以直接作为
DESeq2的输入,执行接下来的分析。 - **ComBat-Seq **使用未标准化的数据作为输入,并且使用负二项回归模型进行批次矫正,ComBat-Seq 使用edgeR,RSEM 的输出数据也OK,但是更推荐使用未标准化的数据作为输入。
- ComBat-Seq 通过比较数据经验分布的分位数与数据中没有批次效应的预期分布来调整原始数据
- ComBat-Seq 可以在R 中获取
GeTMM method
- Smid et al., 2018 提出** GeTMM **(Gene length corrected TMM) 在样本间、样本内的基因表达分析中都具有优势
- GeTMM 基于TMM normalization,但会同时考量 TMM 和 DESeq 或 DESeq2 中缺失的,针对基因长度的Normalize
- GeTMM, 首先计算每个基因的RPK ,然后使用TMM Normalization处理数据,再乘以 1 0 − 6 10^{-6} 10−6进行缩放 (Smid et al., 2018 for detailed calculation)
GeTMM normalization :R package edgeR
load library
library(edgeR)
x <- read.csv("https://reneshbedre.github.io/assets/posts/gexp/df_sc.csv",row.names="gene")
# calculate reads per Kbp of gene length (corrected for gene length)
# gene length is in bp in exppression dataset and converted to Kbp
rpk <- ( (x[,1:6]*10^3 )/x[,7])
# comparing groups
group <- factor(c('c','c', 'c', 't', 't', 't'))
y <- DGEList(counts=rpk, group=group)
# normalize for library size by cacluating scaling factor using TMM (default method)
y <- calcNormFactors(y)
# normalization factors for each library
y$samples
group lib.size norm.factors
ctr1 c 1709962.4 1.0768821
ctr2 c 1674190.8 0.9843634
ctr3 c 1715232.3 1.0496310
trt1 t 1638517.0 0.9841989
trt2 t 1467549.5 0.9432728
trt3 t 935125.2 0.9680985
# count per million read (normalized count)
norm_counts <- cpm(y)
head(norm_counts)
ctr1 ctr2 ctr3 trt1 trt2 trt3
Sobic.001G000200 92.610097 99.192986 68.940090 91.044874 73.623702 93.630285
Sobic.001G000400 5.579741 2.671970 6.172896 2.080457 2.423609 2.547863
Sobic.001G000700 19.324103 27.128459 15.203763 26.026360 34.273836 25.196497
Sobic.001G000800 74.410581 83.143635 71.656315 79.998205 72.089293 75.392505
Sobic.001G001000 9.283023 2.593127 3.164924 2.650027 10.290413 11.014674
Sobic.001G001132 7.464699 4.170389 3.817485 6.392849 9.929718 3.795926
参考
https://www.cnblogs.com/Belter/p/13205635.html
https://www.reneshbedre.com/blog/expression_units.html
https://f1000research.com/articles/4-1521/v1
https://www.jianshu.com/p/35e861b76486