Chem. Eur. J.|针对细胞内靶点的环肽药物:肽药物发展的下一个前沿

题目:Cyclic Peptides as Drugs for Intracellular Targets: The Next Frontier in Peptide Therapeutic Development
文献来源:Chem. Eur. J. 2021, 27, 1487 – 1513
代码:无(环肽综述)
内容:
1.简介
设计可以到达细胞内靶点的大环肽类物质是一个非常大的挑战。本综述主要讨论了合成可以与细胞内靶点接触的环肽设计策略。环肽与传统的小分子不同:因为其特有的物理性质该物质不能评估预测其是否能很好地通过细胞膜。肽合成和实验膜通透性是有效其细胞透膜性好坏的唯一策略。因此,本文主要讨论一些提高细胞透膜性的设计策略:如肽主链N-甲基化以及立体化学变化。然而,这些改进往往是以牺牲生物活性为代价的。因为化学修饰改变了肽的构象,从而影响化合物与目标结合的能力。因此,目前来说侧链的修改是一种更好的方式:即可以提高透膜性也不会太影响结合过程。
2.背景介绍
传统药物的针对靶点的设计会参考以下假设:首先,钟摆转向(pendulum swing)使用组合化学作为开发药物的主要方法,期望所有的目标都可以用一种策略来达到靶点.第二种:第二,认为小而平是药物设计的最佳方法。事实上,新药的设计可比这些简单的假设要复杂得多,因此我们必须使用多种工具设计的针对靶点的药物。其中一个理论就是使用可以体现疾病机制的protein–protein interactions(PPIs)来进行药物发现。以靶点为基础的PPIs要求化合物要在蛋白质上相对大而平的表面(1500–300埃2)牢固并且进行相互作用。然而,一些蛋白质缺乏独特的结构特征,如口袋、裂缝或凹槽,这些结构域往往是小分子调节的结合位点(图1)。
自2000年以来,随着结构和分子生物学技术、x射线晶体学、核磁共振技术和计算建模技术的进步,人们对这些结构有了新的理解。其中一个关键的突破是发现PPIs中蛋白质背后的驱动力并不是均匀地分布在看似大的相互作用表面。涵盖了特定的区域的PPI表面,称为“热点”,这些区域包含了一小部分残基,负责蛋白质之间或蛋白质和配体之间的结合亲和力(图1)。这种热点只占参与相互作用的蛋白质接触面的不到一半,通常出现在接触界面的中心。热点的发现使研究人员能够识别在这些位点上相互作用的分子。
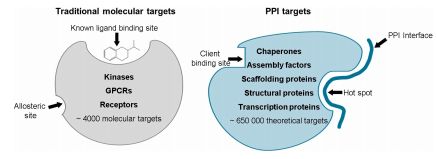
图1 传统分子与PPI靶点的结构特征。
3.作为药物的肽
3.1 肽的介绍
多肽是控制PPIs的理想结构。虽然传统的小分子分子主导了制药市场,但在过去十年中,监管机构批准的肽类药物数量有所增加。与其他类型的药物相比,肽疗法具有许多优势:它们耐受性良好,具有易于插入的立体化学,从而选择性地提供靶标,并且可以与大型蛋白质表面相互作用。肽可以结合那些不稳定但是对于调控PPIs有着重要作用的蛋白质表面,从而具有很好的治疗潜力。目前来说,肽的局限性在于其特有的极性特征。而这种结构会导致肽很难通过细胞膜(脂双层)。
肽结构的改变可以通过被动扩散从而进入细胞。N-甲基化和包含d-氨基酸取代基等修饰策略都可以增加其在细胞的通透性。附着细胞穿透肽(CPPs)也可以通过内吞作用用于肽传递。然而,这些CPPs在化学上是不稳定的,通常会影响肽类药物的结构构象。虽然这些方法可以提高细胞的通透性,但人们经常观察到这些结构修饰会影响在靶标上的结合活性。本文则综述了将多肽转化为细胞渗透性药物导联的方法。
3.2肽:线性还是环形
肽是由酰胺键连接的氨基酸链,由于其广泛的结构多样性,可以构建大的复杂分子,因此是具有吸引力的构建模块。肽药物的范围从寡肽(0-10个氨基酸)到肽(10-50个氨基酸),再到蛋白质(>50个氨基酸)。线性肽作为药物本身是不稳定的,在细胞中被蛋白质水解的可能性很高。这是因为位于两端的游离酸和胺,它们具有典型的极性(图2)。环化促进了环状结构内的分子内氢键结合,降低了分子的外部氢键能力,从而降低了分子的极性,与无环前驱体相比,增加了化合物的膜渗透性。大环肽是通过连接两个原子而产生的,通常位于肽的两端,形成一个环,即环化。环化反应是限制肽构象灵活性的一种方法,成环方式如图2所示。与线性对应肽相比,环肽往往显示出更高的细胞通透性和对蛋白酶的稳定性

图2 典型的肽环化方法包括头对尾、侧链到尾、头对侧链和侧链到侧链。
3.3环肽的优势
环肽具有一定程度的结构预组织,这在线性肽中是没有观察到的。如果环状肽被预先组织成正确的3D构象,与相同的线性肽序列相比,它降低了肽与生物目标结合的熵成本(图3)。大环肽通常具有成为优秀药物的特性;与线性肽相比,它们不仅具有更好的药理特性,而且还提供了通常在小分子中看不到的独特特性。具体来说,大环肽可以从许多包含立体角度的商业起始材料中构建,从而产生结构多样的分子,呈现覆盖与肽序列相关的明确区域的三维空间。强大的固相和溶液相合成方案的发展也使研究人员能够生产几乎任何肽序列,从而覆盖了非常有选择性的空间,包括非天然氨基酸的结合。

图3 线性肽和环状肽的蛋白质目标结合。a)线性肽必须折叠成正确的构象,从而增加结合的熵成本。b)大环结构被预先组织成一个低能量的构象,降低了结合的熵成本。
表1描述了过去10年FDA批准的所有大环肽药物。虽然每年平均有一种药物被批准,但大多数这些分子作用在细胞外靶点。只有一种药物,罗米德蛋白素,成功地与细胞内靶点结合,并于2009年首次被批准用于治疗淋巴瘤。

4.肽的透膜性
4.1透膜性机制
环状肽进入细胞有三种途径:被动扩散、载体介导的转运和内吞作用(图4)。

图4 细胞膜通透性途径包括被动扩散、载体介导的转运和内吞作用。
4.2简介:针对细胞质中靶点的环肽设计
尽管环肽药物具有其独特的优势,但它们通常具有较低的细胞膜通透性。环肽的开发面临的最大挑战之一是确定成功的方法来提高细胞通透性,同时保持生物活性。低细胞膜通透性限制了大多数肽药物抑制位于细胞膜或细胞外基质中的靶点。为了开发环肽药物在细胞内靶点上的优势,我们需要花费大量的精力来开发提高宏观渗透性且不影响环构象的策略。识别适当的预测参数,以准确识别细胞渗透性肽,并通过实验评估修饰肽的渗透性,这是开发新的肽基药物的两种主要策略。
4.3预测大环肽细胞通透性
环状肽的细胞通透性由氢键势、构象、电荷、大小和疏水性等五种特性决定。提高细胞通透性的化学修饰包括改变主链和侧链。虽然使用这些策略对于改善环肽药物的药代动力学特性至关重要,但分子化学结构的任何变化都可能影响其生物活性,必须同时进行评估细胞通透性以及活性。事实上,将肽转化为细胞渗透性分子的研究没有考虑到这些肽药物需要保持其生物活性才能有效。
4.4变色龙假说(The chameleon hypothesis)
人们认为环肽的细胞通透性与它们的动态构象灵活性有关。也就是说,肽根据其周围环境改变其三维形状和疏水性(图5a)。具有这种性质的分子被称为“变色龙”,这解释了为什么尽管具有非常相似的物理性质,一些分子可以进入细胞,而其他分子相对透模性较差。分子变色龙能够调节它们的原子的暴露程度来适应它们周围的环境。为了使大环肽能够具有膜透性,它们必须能够在穿过生物膜时能够采用和交换多种构象。然而,为了使环肽与细胞内靶点相互作用,它必须首先在细胞外采用亲水构象,然后肽必须具有足够的灵活性,在穿过细胞膜时动态切换到非极性结构。一旦进入细胞内的水环境,分子必须重新暴露其极性基团,以保持溶剂化(图5a)。环孢霉素A(CSA)是临床中使用的一种变色龙药物的一个典型例子(图5b)。

图5 环肽的被动通透性与构象的灵活性和“变色龙”行为有关。a)卡通说明变色龙环肽如何在极性和非极性构象之间交换,并通过生物膜扩散。b)环孢霉素A是变色龙药物的一个典型例子,尽管其体积大、高cLogP和高PSA,但它是被动渗透的。
5优化肽大环细胞通透性的化学策略:肽结构的永久修饰
5.1永久修改(permanent modification)
大多数多肽都是被动地通过生物膜扩散的。可以使用几种策略来控制或改变决定细胞通透性的五种特性(氢键电位、构象、电荷、大小和疏水性)。尽管改变可以通过增加侧链和主链的疏水性(即非甲基化)来增加被动扩散,但是主链的改变和侧链修饰通常以不可预测的方式变化。改变也可以提高肽的构象灵活性,使其在通过膜的移动时发生类似变色龙的变化。
其中两个研究最主要的主链修饰是N-甲基化和d-氨基酸的掺入,而侧链的改变通常涉及到非天然氨基酸。虽然这些策略往往会改善环肽的药代动力学,但是任何对生物活性分子的化学修饰都会影响其生物活性。下面描述的是这些策略对改善细胞通透性的有效性及对生物活性的影响的最近研究。
5.2主链N-甲基化
主链N-甲基化是一种化学修饰,通常与改善环肽的药物样性质有关。N-甲基化可以改变环肽的构象、氢键电位和亲脂性,从而提高膜的通透性。在肽主链上添加一个甲基增加了分子的疏水性,并影响了N-甲基化酰胺键的顺式-反式平衡,从而提高了分子作为变色龙的能力。N-甲基化可以对化合物的主链产生长期的影响,这些改变高度依赖于肽序列和手性。因此,使用N-甲基化策略增加亲脂性和改变分子内氢键,从而促进跨细胞膜转运。
虽然在肽主链中引入N-甲基通常可以通过调节构象来改善药代动力学,但N-甲基化也可以影响生物活性。通过改变分子的三维结构,N-甲基化部分的存在可能会影响跟靶点的结合。即使是在肽上添加一个单一的N-甲基化部分,也会引起显著的构象变化,会导致预测该甲基对肽的目标结合亲和力、效价和选择性的影响具有挑战性。尽管存在这一缺点,但主干N-甲基化还是被广泛地应用,因为它显著提高了肽类药物的药代动力学效应。
生长抑素(图6)是一种参与调节内分泌系统的多肽类激素。生长抑素是一种含有14个氨基酸的环状肽,可与位于细胞表面的G蛋白偶联的生长抑素受体结合。生长抑素的一个功能是抑制生长激素;因此,它被用于内分泌疾病的治疗,但由于其药代动力学特性较差,其临床应用有限。一种生长抑素的合成类似物被称为Veber–Hirschmann肽可以解决与生长抑素相关的问题(图6)。Veber–Hirschmann肽保留了生长抑素的活性苯丙氨酸-色氨酸-赖氨酸-苏氨酸序列(蓝色残基如图6所示)。它优先与细胞外五分之二的受体结合,与生长抑素相比,其脱靶效应更少。虽然加入N-甲基化会导致活性下降了4-6倍,但该类似物的结合力仍在低纳摩尔范围内。
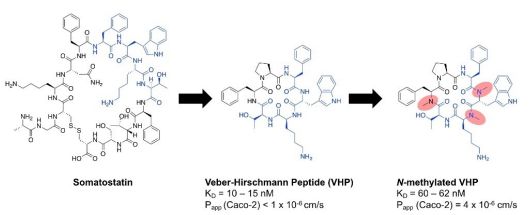
图6 生长抑素、维伯-赫希曼肽和N-甲基化的Veber–Hirschmann肽类似物的结构。对生物活性至关重要的残基用蓝色表示,N-甲基化用红色表示。
Fairlie实验室的同事研究了杂环约束、主链N-甲基化和侧链修饰对血酰胺A细胞通透性的影响(图7)。

图7 天然产物血酰胺A及其丹酰胺衍生物的结构和细胞通透性。主链修饰用红色突出显示,而侧链改变用蓝色圈出。
Masuda及其同事研究了主链N-甲基化对生物活性天然产物PF1171F的影响(图8)。

图8 真菌天然产物PF1171F及其类似物的化学结构、渗透性和生物活性。主链N-甲基化用红色标显示。
诺华公司的Vorherr和他的同事最近进行了另一项关于主链非甲基化对环状肽的影响的研究。他们研究的是多重主链N-甲基化和侧链修饰如何影响环状六肽的细胞通透性和代谢稳定性(图9)。

图9 含有N-甲基化(红色)和侧链修饰(蓝色)的环状六肽及其类似物的结构、渗透性和血浆半衰期。
McAlpine实验室进行的一项类似研究评估了主链N-甲基化对环状五肽LB51的通透性和生物活性的影响(图10)。

图10 环状五肽LB51及其N-甲基化类似物的化学结构、渗透性和结合亲和力。
这些研究表明,主链N-甲基化是一种可合成的策略,可以通过调节大环肽的构象来提高其膜通透性。然而,改变分子的三维结构会影响其与生物目标有效结合的能力。因此,环肽主链N-甲基化作为提高生物活性分子细胞通透性的策略是具有高风险的,如果采用,化合物的药代动力学特性应与生物活性研究同时进行评估。
5.3改变主链立体化学
d-氨基酸的掺入是改善肽类药物性质的一种常见策略,将立体化学从l转化为d可以增加大环肽的膜通透性。一个或多个残基的立体化学反转改变了分子的三维构象,从而影响了大环肽药物的整体疏水性、两亲和性和变色龙行为。除了图11所示的PF1171F的主链N-甲基化研究外,Masuda和同事还研究了修饰主链立体化学的影响。

图11 真菌天然产物PF1171F及其类似物的化学结构、渗透性和生物活性。立体化学修饰用绿色突出显示。
除了评估主链N-甲基化对LB51的影响(图10),McAlpine实验室还研究了结合d-氨基酸生成该先导分子的非对映体的影响(图12)。

图12 环状五肽LB51及其非对映体的化学结构、渗透性和生物活性。
与N-甲基化的结果一致,通过改变主链立体化学,可以通过改变环肽的构象来增加它们的通透性。然而,对生物活性化合物的三维结构的修饰会影响它们有效地与目标结合的能力。因此,虽然改变环的立体化学是提高生物活性分子的细胞通透性的有用策略,类似于主链非甲基化,但它是一种高风险的方法,可能对生物活性产生不可预测的影响。因此,这些化合物的药代动力学特性应在研究其生物活性的同时进行评价。
5.3修饰大环肽骨架的新策略
Hickey和来自Yudin实验室的同事研究了将外部酰胺加入到环肽中作为增强其渗透性的一种策略(图13)。

图13 外环酰胺作为调节环肽构象和提高被动渗透性的策略。
Wu和Kodadek实验室的同事研究了将环丙氨酸作为生物活性环肽的非甲基化方法的替代效果(图14)。

图14 将环丙氨酸加入大环多肽是提高细胞通透性的一种很有潜力的策略。
与环丙氨酸修饰类似,Matsui和同事研究了将环丙烷链作为另一个环主干部分的影响(图15)

图15 环丙烷系链可用于控制大环多肽的构象,以增强细胞的通透性。
Verma和他在查特吉实验室的同事研究了将硫酰胺纳入生物活性环肽西伦吉地肽的主链的影响(图16)。
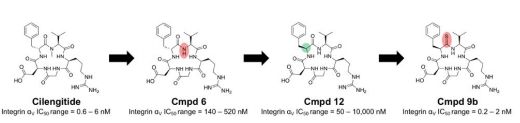
图16 在主碳基中加入硫取代氧是控制大环肽构象的有效策略。
Appavoo和Yudin实验室的同事评估了另一种修改大周期主干的新策略。从天然产物如血酰胺A(图7)中获得灵感,恶二唑被纳入到长度为4到8个氨基酸的模型环肽的主干中。这种修饰稳定了这些肽在DMSO和水中的溶液构象,其中水环境中的结构更具有生物学相关性。主链杂环加强了分子内的氢键模式,以稳定分子的三维结构。鉴于通过测试肽获得的有希望的数据,我们将该策略应用于生物活性支架(图17)。

图17 将杂环作为肽主干的内环控制元件,可作为提高渗透性的策略,同时保持生物活性。
McAlpine和他的同事对三硫酰胺A的衍生物进行了研究(化合物a,图18)。
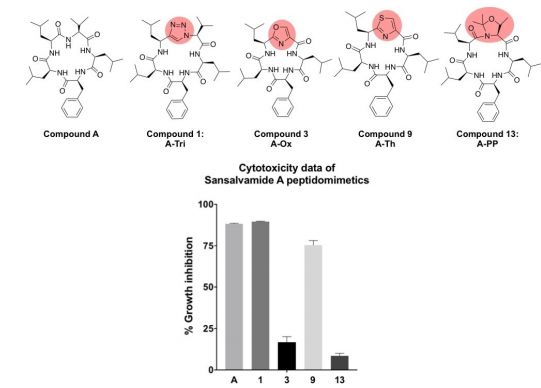
图18 将杂环并入肽主干作为结构控制元件可用于维持渗透性和生物活性。
5.4永久侧链修改(Permanent sidechain modifications)
修饰肽的侧链是一种可以用来提高其生物活性和药代动力学,特别是膜通透性的策略。侧链修饰通常包括合并非天然氨基酸,其中侧链连接到主链氮原子,而不是a-碳(图19)。这些变化会干扰对蛋白水解酶的识别,并增加肽基药物的半衰期。非天然氨基酸包括疏水基团,如烷基链,它增加了肽的亲脂性,通常与增强的渗透性有关。这种侧链修饰可能通过改变侧链到主链的相互作用来改变肽的构象,从而极大地影响膜的通透性和结合亲和力

图19 具有侧链修饰的CXCR7调节剂的结构及其对药理学和膜渗透性的影响。
5.5掩蔽或前药部分(Masking or prodrug moieties)
肽的细胞通透性也可以通过使用不稳定的产生前药物衍生物的极性侧链基团来提高。前药设计的基本原理是,引起预期药理作用所需的结构成分和膜转运和传递到分子靶点所需的结构成分并不相同。前部分是不稳定的保护基团,有助于前药细胞的通透性,但在进入细胞环境时可以从肽中被酶裂解(图20)。有许多酶活性不稳定的前部分可以帮助跨膜转运,包括磷酸盐、醚、酯、碳酸盐和酰胺。利用这种可逆衍生化技术,获得具有增加亲脂性、代谢稳定性和提高细胞通透性的问题肽的衍生物是可行的。

图20 应促进细胞通透性的掩蔽组策略。
McAlpine实验室之前的研究研究了d-氨基酸和n-甲基对极性分子LB51的影响。为了评估侧链修饰对该支架的影响,他们在亲水侧链上加入了甲基,以掩盖其极性(图21)。

图21 环状五肽LB51及其甲基化衍生物的化学结构、渗透性和生物活性。
舒马赫-霍夫曼实验室以及凯斯勒和霍夫曼实验室的同事研究了用疏水前药部分掩盖亲水性残基对一系列环六肽的细胞通透性的影响(图22)。
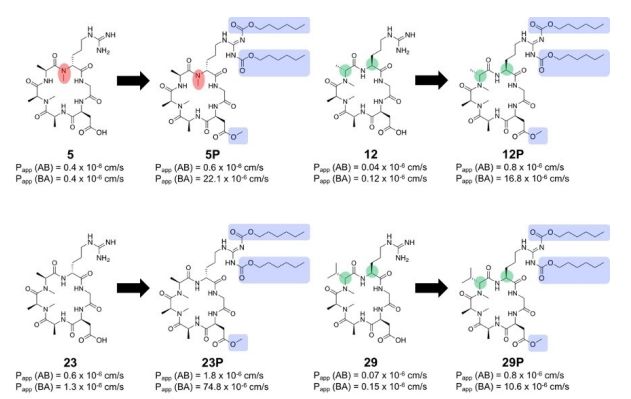
图22 亲脂性掩蔽基团可作为提高极性和带电环肽的渗透性的策略。双向Caco-2渗透率值显示在每个化合物的下面。
4总结展望
开发能够与细胞内靶点结合的大环肽是一个重大的挑战。我们需要了解一个分子需要识别并结合位于细胞内的目标,并随后引发生物反应。计算出的物理性质不能充分预测复杂分子变色龙的细胞渗透性,如大环肽。预测变色龙行为的计算模型正在出现,随着这些方法变得更加复杂,所产生的数据可能有助于设计或工程有效的细胞渗透性环肽。包括主链N-甲基化和立体化学变化在内的化学策略拓宽了我们对结构多样的3D分子如何进入细胞并产生具有提高通透性的分子支架的理解。然而,这些改进往往是以牺牲生物活性为代价的。这种侵入性的化学修饰改变了环肽的构象,会影响化合物与细胞内目标结合的能力。很明显,构象限制是与生物目标紧密结合亲和力的一个重要因素,减少分子的构象空间将减少可用构象的数量,产生有效的化合物。然而,柔韧性和变色龙行为对膜的渗透途径至关重要。
变肽主链或掩蔽极性侧链的新兴策略似乎是开发治疗相关的大环肽的有效方法。本文的综述表明,在不同检查结合亲和力的情况下优化膜的渗透性不一定有效,这些特性应该同时优化。新的方法旨在开发极性侧链的临时掩蔽基团,其中这些基团可以在细胞内被裂解。这种方法在设计未来的极性肽药物时可能有效。因为在进入细胞的过程中,临时掩蔽基团隐藏了极性侧链,但一旦在细胞质中就释放活性化合物。因此,临时掩蔽基团允许膜的通透性,而不影响结合亲和力。将这种新方法应用于未来的肽药物将改变药物开发计划,使肽成为主流药物分子,用于靶向细胞内蛋白质。事实上,这种方法应该会有效促进肽类药物的发现。