单细胞转录组实战04: infercnvpy识别恶性细胞
 生信交流与合作请关注公众号@生信探索
生信交流与合作请关注公众号@生信探索
Infercnv is a scalable python library to infer copy number variation (CNV) events from single cell transcriptomics data.
Input data
adata.X 数据需要标准化和log转化,包括全部基因。
下载GENCODE 基因注释信息

mkdir -p ~/DataHub/Genomics/GENCODE/release_42
cd ~/DataHub/Genomics/GENCODE/release_42
# 下载参考基因组及注释文件
#>>>downGENCODE.sh
wget https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_42/gencode.v42.annotation.gtf.gz
mv gencode.v42.annotation.gtf.gz HS.gencode.v42.annotation.gtf.gz
#<<
nohup zsh downGENCODE.sh &> downGENCODE.sh.log &
导库
from pathlib import Path
import pandas as pd
import scanpy as sc
import infercnvpy as cnv
import matplotlib.pyplot as plt
OUTPUT_DIR='output/03.inferCNV'
Path(OUTPUT_DIR).mkdir(parents=True,exist_ok=True)
adata_raw = sc.read_h5ad('output/adata.h5').raw.to_adata()
CellTypeKey="CellTypeS2"
CNVCellTypeKey="CellTypeS3"
为adata.var添加基因信息
每个基因的chromosome, start, and end
cnv.io.genomic_position_from_gtf("~/DataHub/Genomics/GENCODE/release_42/HS.gencode.v42.annotation.gtf.gz",adata_raw)
infercnv
基因按照染色体和基因组位置排序;与reference比较基因组区域的基因平均表达水平。
reference是提前知道的正常细胞,官网用全部的免疫细胞做reference
window_size:window里边的基因数目,基因多点好
这一步添加cell x genomic_region 矩阵到 adata.obsm['X_cnv']
大概计算过程,有助于理解各个参数:
- 计算logFC
adata.X中的数据已经log转化过,所以,这一步的减法相当于计算logFC
所有细胞减去reference的基因表达量。
多个reference_cat的计算方法,means=各个reference平均值就是分开算reference_cat里每个reference表达量的平均值;若表达量在means的最小值和最大值内,则logFC=0;若表达量小于means的最小值,则表达量-该最小值;若表达量大于means的最大值,则表达量-该最大值。
- Clip the fold changes at
-lfc_capand+lfc_cap. - 计算每10(
step)个window内的基因表达量,window为100(window_size)个基因; - 中心化,每个细胞减去该类细胞的中位数;
- 噪音过滤,数值小于
dynamic_theshold * STDDEV则=0,STDDEV是基因的标准差。 - Smooth the final result using a median filter.
np.unique(adata_raw.obs[CellTypeKey])
cnv.tl.infercnv(
adata_raw,
reference_key=CellTypeKey,
reference_cat=['B', 'CD4+ T', 'CD8+ T', 'DC', 'Mast','Treg', 'pDC','Plasma'],
window_size=250
,n_jobs=12
)
各类细胞在染色体上的基因表达量
cnv.pl.chromosome_heatmap(adata_raw, groupby=CellTypeKey)

细胞聚类
和细胞注释时的聚类类似,只是使用的数据时adata.obsm['X_cnv']中的
包括3步算PCA、算距离neighbors、leiden聚类
各cluster在染色体上的基因表达量图,前几个cluster没有显著差异的表达的基因组区域。显著差异的表达的基因组区域可能是由于CNV,因此这些cluster可能是肿瘤细胞。(前几个cluster可能是肿瘤细胞)
cnv.tl.pca(adata_raw)
cnv.pp.neighbors(adata_raw)
cnv.tl.leiden(adata_raw)
cnv.pl.chromosome_heatmap(adata_raw, groupby="cnv_leiden", dendrogram=True);

CNV分数
Clusters有高的分数说明其更可能受到CNV影响,更可能是肿瘤细胞。
9、23分数较高而且没有聚类在一起,可能是肿瘤细胞。
score=mean of the absolute values of the CNV matrix for each cluster.
cnv.tl.umap(adata_raw)
cnv.tl.cnv_score(adata_raw)
_, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(18, 6))
cnv.pl.umap(
adata_raw,
color="cnv_leiden",
legend_loc="on data",
legend_fontoutline=2,
ax=ax1,
show=False,
)
cnv.pl.umap(adata_raw, color="cnv_score", ax=ax2, show=False)
cnv.pl.umap(adata_raw, color=CellTypeKey, ax=ax3,show=False);
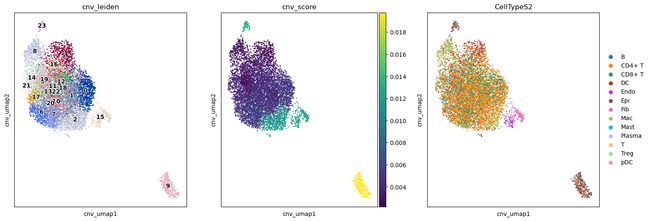
观察CNV分数在各类细胞中的分布。
_, (ax1, ax2, ax3) = plt.subplots(1, 3, figsize=(18, 5), gridspec_kw=dict(wspace=0.5))
sc.pl.umap(adata_raw, color="cnv_leiden", ax=ax1, show=False)
sc.pl.umap(adata_raw, color="cnv_score", ax=ax2, show=False)
sc.pl.umap(adata_raw, color=CellTypeKey, ax=ax3, show=False);

根据以上结果把9和23判断为肿瘤细胞。
adata_raw.obs["cnv_status"] = "Normal"
adata_raw.obs.loc[adata_raw.obs["cnv_leiden"].isin(['9','23']), "cnv_status"] = "Tumor"
adata_raw.obs["CellTypeS3"] = adata_raw.obs[CellTypeKey].tolist()
adata_raw.obs.loc[adata_raw.obs["cnv_leiden"].isin(['9','23']), 'CellTypeS3'] = "Tumor"
fig, (ax1, ax2,ax3) = plt.subplots(1, 3, figsize=(18, 5), gridspec_kw=dict(wspace=0.4))
cnv.pl.umap(adata_raw, color="cnv_status", ax=ax1,show=False)
sc.pl.umap(adata_raw, color="cnv_score", ax=ax2,show=False)
sc.pl.umap(adata_raw, color="cnv_status", ax=ax3,show=False);

保存数据
adata = adata_raw.copy()
adata.raw = adata
adata = adata[:,adata.var.highly_variable]
adata.write_h5ad(OUTPUT_DIR+'/adata.h5',compression='lzf')
Reference
https://icbi-lab.github.io/infercnvpy/tutorials/tutorial_3k.html
本文由 mdnice 多平台发布