Nature Communications|评估推进基于网络的蛋白质-蛋白质相互作用预测的社区工作

题目:Assessment of community efforts to advance network-based prediction of protein–protein interactions
文献来源:Nature Communications | (2023) 14:1582 4
代码:https://github.com/spxuw/PPI-Prediction-Project
内容:
全面了解人类蛋白-蛋白相互作用(PPI)网络,也就是人类相互作用组,可以为了解复杂的生物过程和疾病的分子机制提供重要的见解。尽管迄今为止,在确定人类相互作用组的结构方面进行了显著的实验努力,但许多PPIs仍未被绘制出来。计算方法,特别是基于网络的方法,可以促进以前未确定特征的PPIs的识别。人们已经提出了许多这样的方法。然而,对现有的基于网络的PPIs预测方法仍缺乏系统的评价。在这里,作者报告了由国际网络医学联盟发起的社区工作,以衡量26种具有代表性的基于网络的方法预测四种不同生物体的6个不同相互作用组的PPIs的能力:A. thaliana, C. elegans, S.cerevisiae, and H. sapiens。通过广泛的计算和实验验证,他们发现先进的基于相似度的方法,利用了PPIs的底层网络特征,在所考虑的反应组中显示出优于其他一般链路预测方法的性能。
1.背景介绍
对人类PPI网络(也被称为人类相互作用组)的全面了解可以增加对细胞组织、基因组功能和基因型-表型关系的全面见解。对未确定特征的PPIs的发现可以促进重要的干预目标,如药物靶点的识别和治疗设计。尽管在高通量方面付出了显著的实验努力,但人类交互组图仍然不完整,并受到噪声和调查偏差的影响。这些因素严重地阻碍了准确理解细胞组织和基因组功能。计算方法可以通过显著减少实验中确认的替代方案的数量,加速生物医学网络中的知识获取。然而,人类交互组图的高度不完整性可能会降低最先进的计算方法的有效性。在这种情况下,基于实验观察到的PPIs对先前未确定特征的PPIs进行计算预测成为一项特别具有挑战性但可能具有高回报的任务。
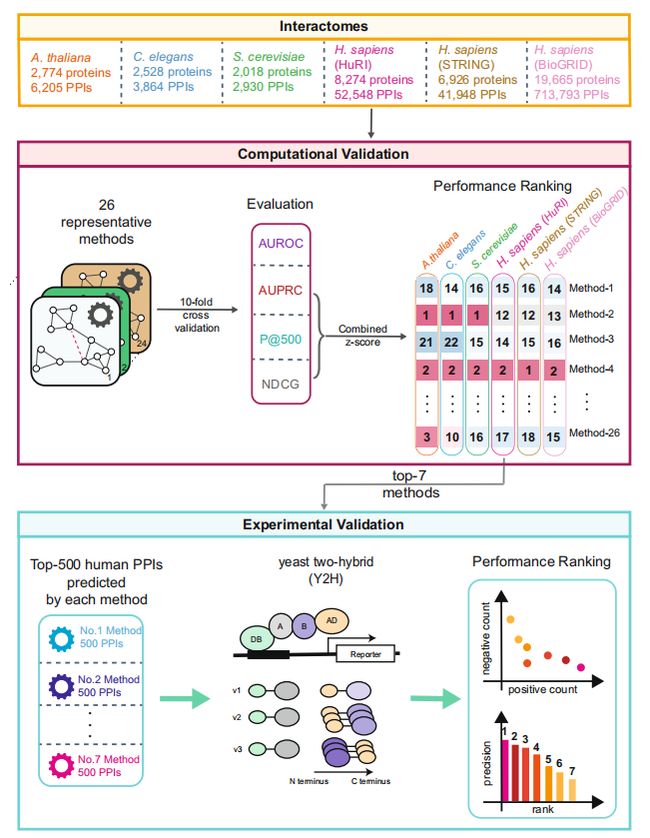
图1 INMC PPI预测项目的工作流程。系统评估了26种具有代表性的基于网络的方法来预测四种不同生物体相互作用组中的PPI:A. thaliana, C. elegans, S.cerevisiae, and H. sapiens.:HuRI4, STRING和BioGRID(使用rTRM软件包)。在计算验证过程中,通过10倍交叉验证,将每个交互组的PPIs分为训练集和验证集。每种方法的性能都使用四个标准指标进行评估:AUROC,AUPRC,P@500,NDCG。对于每种方法,一个总体得分被定义为三个指标的z分数的总和((AUPRC,P@500和NDCG)。根据其在计算验证过程中预测人类PPIs的性能,选择了前7种方法。使用整个人类相互作用组,前7种方法都预测了前500个人类PPIs,并使用Y2H试验进行实验验证。蛋白质间的相互作用。AUROC:接收机工作特性曲线下的面积。AUPRC:精度-召回曲线下的面积。P@500:前500个预测PPIs的精度。NDCG:标准化的折现累积增益。Y2H:酵母双杂交试验。测定方法1-测定方法3。
在PPI预测的环境中,认识到不同的计算方法的优点和局限性是至关重要的,这为选择最佳的预测策略提供了所需的见解。为了加速这一过程,国际网络医学联盟(INMC)启动了一个项目,通过标准化的性能指标和标准化的、无偏向性的交互组分析,系统地对26种具有代表性的基于网络的PPI预测方法进行基准测试(该项目的工作流程总结见图1)。26种方法的缩写和简要描述见表1。该工作涵盖了链路预测的主要类别,从基于相似性的方法到概率方法、基于因素分解的方法、基于扩散的方法和基于机器学习的方法(图2)。需要注意的是,在26种基于网络的方法中,除了有3种基于PPI预测的拓扑信息外,还利用了生物数据(即蛋白质的序列信息)。
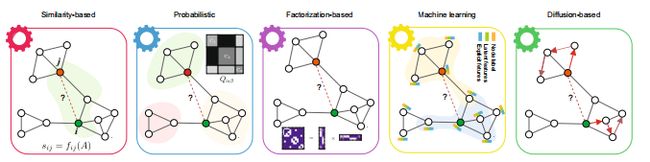
图2 五大链路预测方法的示意图。(1)基于相似度的方法。这些方法基于图中节点之间预定义的相似性函数,即共同的邻居(绿色区域)的可能性来量化链路的可能性。(2)概率性的方法。这些方法假设真实的网络有某种结构,如社区结构。这些算法的目标是选择模型参数,可以最大化的可能性的观察结构。群落内节点的连接概率高于不同群落间节点的连接概率(灰色矩阵)。(3)基于因式分解:这些方法的目标是通过保留全局网络模式来学习图中每个节点的低维表示。接下来,通过计算相似度函数或训练分类器,利用压缩表示来预测未观察到的PPIs。(4)机器学习:在机器学习类别中有很多方法;在这里,我们使用最先进的图神经网络(GNN)来说明这个类别。这些方法通过利用神经网络聚合节点特征、链路特征和图结构,并通过图中的链路传递信息来嵌入节点信息。然后,学习到的表示被用来训练一个监督模型来预测缺失的环节。(5)基于扩散:这些方法使用的技术是基于分析从网络上的随机步行的扩散运动中收集到的信息(路径由红色箭头表示)。

表1 在INMC PPI预测项目中测试的计算方法。
为了评估这些方法的性能,我们需要可靠和无偏不倚的基准相互组。文献中准备的的具有多个支持证据线的PPIs相互作用组可能是高度可靠的,但它们在很大程度上受到选择偏差的影响。因此,在这里,作者关注的是从缺乏选择偏差的系统筛选中出现的相互作用组。为了简单起见,主要关注不包含共复杂成员注释的二进制数据集。以下6个基准相互作用组进行用于性能评价:(1)一个植物相互作用组,包括2774个蛋白质和6205个PPIs,来自于PPIs A. 拟南芥相互作用组(A. thaliana Interactome);(2)一个蠕虫的相互作用组,包括2528个蛋白质和3864个PPIs,来自于 C. 秀丽隐杆线虫第8版(C. elegans version 8 (WI8);(3)酿酒酵母酵母相互作用组,包括2018个蛋白和2930个PPIs,来自CCSB-YI1、Ito-core和uetz筛选数据集;(4)一个包括8274个蛋白质和52548个PPIs,来自HuRI,它是由三个独立的高质量Y2H筛选的二元蛋白相互作用组装而成。为了确保评估的普遍性,还使用两个额外的来自不同方法的相互作用组:(5)人类相互作用组包括6926蛋白质和41948物理PPIs,来自STRING过滤后PPIs标准化分数低于0.9的高置信PPIs和(6)人类相互作用组包括19665蛋白质和713793物理PPIs,来自BioGRID。
对于这6个交互组中的每一个,作者都进行了10倍交叉验证,以评估26种不同方法的4个性能指标(在这里我们将这个过程称为“计算验证”)。这种分析允许这些方法进行排序。接下来,根据它们在预测人类交互作用组中相互作用方面的表现选择前7种方法,并选择其预测的前500个人类PPIs(累积3276个PPIs)通过Y2H分析进行系统和无偏的实验验证。总的来说,我们验证了1177个以前未被鉴定的PPIs,涉及633个人类蛋白。在文献中没有其他基于联盟的PPI预测算法的评估纳入了如此大的实验验证工作。
在本文中,作者报告了这一社区工作的结果,其中评估了在PPI预测环境中各种链路预测算法的性能,并为检测未映射PPI所需的最优计算工具提供了见解。作者发现,先进的基于相似性的方法,利用了PPIs的潜在特征,在所考虑的交互作用组的计算和实验验证中,其结果优于其他预测方法的性能。作者描述了这些方法的细节。完整的数据集(包括所有的基准交互组和实验验证结果),以及所有的测试方法和评分函数的代码,都可以免费提供给科学界。
2.结果
2.1 不同性能指标之间的相关性
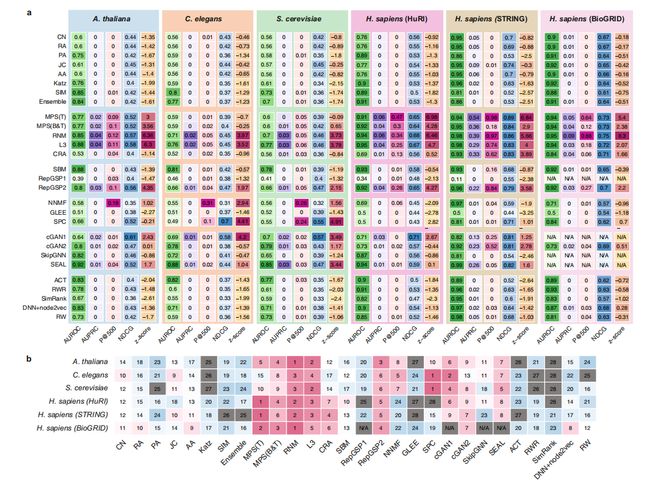
图3 PPI预测方法的计算指标。每种方法的细节总结在表1中。a 一个热图图显示了每种方法在每个相互作用组上的性能,并具有以下评价指标:AUROC、AUPRC、P@500和NDCG。总体性能是由三个指标的z分数计算出来的。对于每个指标,较深的颜色代表更好的性能。b 26种方法在6个交互组中按z分数进行的排序。需要注意的是,由于计算成本高昂,ReGSP1、cGAN1、SEAL和SkipGNN在BioGRID数据库上的性能没有得到评估。我们把他们的排名标记为无排名。请注意,在计算每种方法的组合z分数和排名时,都排除了AUROC。
图3 总结了所有使用10倍交叉验证的测试方法的计算评估结果。用于量化每种方法性能的指标为(i) AUROC:接收机工作特征下面积;(ii)AUPRC:精度召回曲线下面积;(iii)NDCG:归一化贴现累积增益;(iv)P@500:正PPIs的比例,即前500个预测中的精度。为了更好地对不同方法的总体性能进行排序,作者还计算了每种方法的组合z分数,该方法使用不同的评估指标来总结其性能。接下来,强调以下从分析中得到的关键观察结果和见解。通过各种PPI预测方法,发现AUROC在很大程度上高估了任何特定方法的性能。例如,在H.sapiens(HuRI)中,SEAL的10倍交叉验证的平均AUROC为0.94。这个非常高的AUROC可能会导致我们错误地得出结论,认为SEAL是一个几乎是完美的预测方法,因为AUROC的最大值是1。然而事实上,SEAL的平均AUPRC只有0.012,这意味着在寻找PPIs方面表现较差(图3a)。由于AUROC在预测文献中得到了广泛的应用,作者研究了基于AUROC的链路预测方法的排名是否与基于AUPRC或其他指标的排名相一致。首先,计算了所有方法中AUROC和其他指标之间的相关性。AUROC与AUPRC(Spearman R = 0:75,p < 2:2×1016)和NDCG(Spearman R = 0:76,p = 1:6×1014)显著相关,但与P@500(Spearman R = 0:18,p = 0:028)没有显著相关。其次,作者发现不同方法的组合z分数,包括和排除AUROC度量,是相当一致的(Spearman R = 0:97,p < 2:2×1016,见补充图1d)。请注意,AUROC和AUPRC之间的强相关性并不意味着前者在评估任何特定链路预测方法的性能方面与后者一样好。相反,这只是意味着我们仍然可以使用AUROC来对不同的方法进行排名(排名将和我们使用AUPRC一样大致相同),即使AUROC值被系统地膨胀了(由于数据不平衡的问题)。因此,在计算组合z分数以及对每种方法的性能进行解释时作者排除了AUROC。
2.2 相互作用组的可预测性较弱
值得注意的是,除了H. sapiens (STRING)外,大多数方法对5个相互作用组都很小(图3a)。这一观察结果表明,成功地预测一个大的未映射的PPI空间之间的缺失环节仍然是一项具有挑战性的任务。为了量化每个相互作用组的可预测性,基于相互作用组的邻接矩阵的一阶扰动计算了其结构一致性指数σc。需要注意的是,如果移除或添加一组随机选择的链接并没有显著改变网络的结构特征(以其邻接矩阵的特征向量为特征),那么网络是高度可预测的(具有高σc)。结果显示这可能意味着H. sapiens (STRING)可能是最没有偏向性的,其他相互作用组的低结构一致性表明了五个相互作用组中未观察到的部分本与目前观察到的部分没有非常相似的结构特征,这可能是由于这些相互作用组图的高度不完整性。
2.3 大多数PPI预测方法的性能在不同的交互作用组中有很大的差异
H. sapiens (STRING)的PPIs(HuRI和STRING)比其他三种生物的PPIs预测更准确,尽管它们的边缘密度非常相似。为了更好地证明不同方法的性能可变性,作者根据AUPRC、P@500和NDCG的组合z分数对这些方法进行了排序。如图3b所示,一些方法(如RNM)在不同的相互作用组中表现出相当稳定的排名(或一致的性能),而其他方法的排名在相互作用组中显示出非常大的可变性。这种可变性可能是由于不同的网络特征导致的,即链路的数量和平均程度。反过来,这些在一定程度上取决于现有PPIs的完整性程度,所以导致在不同的相互作用组中可能有很大的差异。为了系统地评估平均性能与可变性,作者分别计算了每种方法的所有6个交互组的排名的平均值和标准差,发现RNM产生了最高的平均排名和最低的可变性。这表明RNM具有鲁棒性,能够在具有完全不同网络特征的交互体上表现良好。
2.4 传统的基于相似度的方法表现不佳
传统的基于相似度的关系预测方法在基准研究中已被广泛应用。这些方法基于一系列简单的节点相似度得分,如共同邻居的数量、Katz指数、Jaccard指数和资源分配指数,以量化潜在关系的可能性(见表1)。因此,它们比其他方法更容易被解释和可扩展。然而,预定义的局部和全局相似度评分函数可能会显著影响其对某些网络的预测能力,如PPI网络。例如,仅仅在一个相互作用组中存在两个蛋白质并不一定意味着它们应该相互连接,因为相互作用的蛋白质不一定是相似的,相似的蛋白质也不一定是相互作用的。作者发现,大多数传统的基于相似性的方法,即common neighbors (CN), Adamic-Adar index(AA), and Jaccard index (JC), yield negative z-scores,产生的z分数为负,表明它们的性能低于所有方法的平均性能(图3a)。
2.5 有六种持续的高性能的方法
在测试的所有方法中,RNM、L3、MPS (T)、MPS(B&T)、RepGSP2和SEAL在6个交互作用组的计算评估中产生了相对较高的AUPRC和P@500,具体例子可见原文。
2.6 在每个交互作用组中,堆叠模型的表现并不比明显优于单个方法
以前已经有人提出,构建一系列的“堆叠”模型,并将它们组合成一个单一的预测算法,可以达到最优或接近最优的精度。为了确定堆叠模型在PPI预测方面是否优于个别方法,作者构建了四种不同的堆叠模型。堆叠模型1(监督):堆栈36个拓扑预测器,分为三种类型,全局(整个网络的功能),成对的节点对ij)和基于节点(节点i和节点的独立拓扑属性的函数和j),到一个单一的算法,然后训练一个分类器来预测缺失的链路。堆叠模型-2(无监督):对于每个链路,取根据RNM和MPS(T)计算的排名平均值(百分位形式)。堆叠模型-3(无监督):对于每个链路,取根据RNM和MPS (T)计算出的最大排名。堆叠模型-4(无监督):对于每个链路,使用RNM或cRanl方法近似的Kemeny共识40聚合RNM和mans(T)的排名。有趣的是,这些堆叠模型中没有一个能显著优于单个方法。一般来说,堆叠模型-1的优点是元分类器可以通过监督训练学习选择最佳预测器;因此,堆叠模型的整体性能优于任何单个预测器。然而,在案例中,两种排名最高的方法的预测因子可以直接用于PPI预测,而无需训练一个分类器。此外,不同方法预测的PPIs之间的重叠程度很低。因此,简单地平均不同方法的分数(stack模型-2)会降低正确预测的PPIs的排名,从而相应地降低预测性能。堆叠模型-3可以产生略高的AUROC。但其AUPRC和P@500仍不能超过最好的单个方法。这些结果表明,对于一个给定的网络域(如PPI网络),堆叠模型在链路预测中并不总是显著优于最好的单个方法。
2.7 预测PPI的模式
基于在预测H. sapiens (HuRI)相互作用组的PPIs方面的表现,选择了前7种方法:MPS (T)、RNM、MPS(B&T)、cGAN、SEAL、SBM和DNN+node2vec(。为了检验前7种方法预测的前500个PPIs中是否存在特定的模式,作者计算了预测的PPIs中涉及的蛋白质的程度差异分布(图4a)。RNM、MPS (T)、MPS(B&T)和SBM倾向于预测蛋白质程度更高的的PPIs。其余的方法cGAN、SEAL和DNN + node2vec倾向于预测具有相似程度的节点之间的PPIs。此外,对于每种方法,我们在人类相互作用组的度分布P(K)之上(平均度为12.7),绘制了参与其前500个预测PPIs的蛋白质的平均度(图4b)。MPS(B&T)倾向于预测涉及高度蛋白质的PPIs,而深度学习方法如DNN节点节点2vec和+等前500个PPIs中蛋白质的平均程度要低得多。这种差异可能是由于RNM为高级节点提供了更多的预测(见图4b),MPS也在高水平上利用了L3原理,而DNN + node2vec更关注局部网络拓扑结构而不是度。
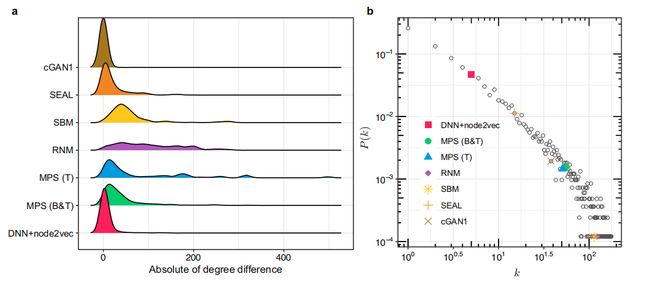
图4 前7种人类PPI预测方法预测的前500个PPI的模式。a对于这些前七种方法,作者检查了每个蛋白质对的程度之间的绝对值的分布。bH. sapiens (HuRI)相互作用组的程度分布和在对数-对数图中每种方法预测的前500个PPIs中参与的蛋白质的平均程度。K表示蛋白质的程度。
2.8 预测方法在实验验证中的性能
据作者所知,该工作是一个系统基准研究中对基于网络的PPI预测方法的一个前所未有的大规模实验验证。为了实验验证PPI预测方法的性能,他们将前7种方法(MPS (T)、RNM、MPS(B&T)、cGAN、SEAL、SBM和DNN + node2vec)应用于人类交互组(HuRI),并实验验证了预测的前500个未映射的人类PPI。从前7种方法中预测的前500个人类PPIs的结合包括3276对独特的蛋白质对。接下来,作者使用以前用于获得HuRI图的三种正交Y2H分析来验证这些蛋白质对。预测的PPIs集的总体恢复速度与文献中高可信的二元PPIs的恢复速度相当。总之,作者确定了1177以前未确定人类PPIs(涉及633个蛋白质)。请注意,由于技术问题,一些蛋白质对没有被成功测试。总的来说,MPS(B&T)是最有前途的方法,在其预测的前500个PPIs中,它同时提供了最高的阳性对(376)和最低的阴性对(54),精度为87.4%(见图5)。另外两种很有前途的方法分别是MPS (T)和RNM,其精密度分别为75.9%和69.5%。需要注意的是,在计算验证中,MPS(B&T)排名No.3(根据预测人类PPIs的组合z分数),而MPS(T)排名No.1,RNM排名No. 2。在计算和实验验证方面的排名差异并不大,令人惊讶。

图5 前7种人类PPI预测方法的实验评价。如果一个蛋白对在三种Y2H检测中至少一种为阳性,则认为为阳性;如果在三种检测中均为阴性,则认为为阴性。MPS(B&T)是最有前途的方法,在预测的前500个PPIs中,同时提供阳性对(376)和阴性对(54),精度为87.4%。请注意,未成功测试的蛋白质对的数量(例如,由于移液失败)不包括在精度计算和此图中。
2.9 结合前三种方法的预测并不能产生更好的精度
排名较高的预测PPIs(即在前500名的顶部)可能比那些排名较低的预测PPIs(即在前500名的底部)在实验验证中具有更高的阳性概率更高。为了验证这一假设,对于人类PPI预测的前三种方法,作者绘制了Y2H实验中被验证为阳性的PPI的排名位置分布。令人惊讶的是,这些积极的PPIs并不会更频繁地出现在列表的顶部。相反,它们几乎是随机地出现在每种方法预测的前500个PPIs中。目前还不清楚,如果测试更多的测试对,比如前1000个ppi,这种有趣的现象是否会继续存在。)因此,将这些顶级方法预测的前500名PPIs结合到一个新的前500名列表中,在实验验证中并不能产生更好的性能。
2.10 已验证的和以前未确定特征的人类PPIs的结构和功能关系
为了探索这些在Y2H检测中检测为阳性的预测PPIs的结构关系,可视化了由它们构成的网络(总共1177个PPIs,涉及633个蛋白),发现了4个不同的簇(图6)。这些聚类主要由RNM、SBM和MPS方法贡献。由RNM和SBM贡献的子网络非常接近。MPS(B&T)和MPS (T)共同形成一个集群。此外,MPS(B&T)还对集群本身做出了贡献。作者还研究了这些阳性PPIs的功能关系,发现它们参与了三个功能模块,并且每个功能域都与一个独特的、丰富的GO相关。.这一观察结果也与之前的发现相一致,即物理结合可以将蛋白质组装成大的功能群落,从而为了解人类细胞的整体功能组织提供了思路。具体来说,作者发现参与前500个预测PPIs的蛋白参与了表皮,如角质化、角蛋白丝和角化,这也适用于实验验证的阳性PPIs。

图6 以前未确定特征的人类PPIs之间的结构关系。该网络包括所有1177种以前未确定特征的人类PPIs,由前7种方法预测,并通过Y2H分析进行验证。用单一方法预测的PPIs根据预测它们的方法着色。用多种方法预测的PPIs(即在预测的前500个PPIs中)用黑色表示,边缘宽度与预测该PPI的方法数量成正比。节点(蛋白质)的颜色是基于它们所属的连接组件组成的。节点大小与节点程度成正比。值得注意的是,共有174个分离的节点,代表自相互作用的蛋白(主要由cGAN1检测到)。
3 讨论
在这个项目中,作者专注于对26种基于网络的方法进行基准测试。在这26种方法中,有3种方法(即MPS(B&T)、SEAL和RW2)也利用了蛋白质序列信息。作者已经意识到那些纯粹基于序列的PPI预测方法(例如,SVM45-48,RF49,50,FCTP51和DPPI52),以及这些方法利用了额外的生物信息,如3D蛋白质结构和蛋白质注释。我们在这个基准研究中没有考虑这些方法,有两个原因。首先,这些方法需要为每个链路定义一个特性空间,这将导致HuRI(拥有~3500万未映射PPI)带来显著的时间复杂度和内存需求。系统地对这些方法进行基准测试只是超出了当前项目的范围。其次,结构信息对构建相互作用组的影响相对较小,主要是因为已知序列的蛋白质数量与实验确定的三级或四级结构的蛋白质数量有很大的差异53。换句话说,这类信息太不完整了,无法被有效地利用。
基于这些以上发现,作者建议提出有效预测PPI的以下考虑因素。首先,该方法需要利用相互作用组的固有特性(如L3原理)来提高预测性能。其次,未映射的PPI空间比当前映射的空间大几百倍以上,导致不同方法预测的最可能的PPI空间的有限重叠,明显降低了集成或堆叠模型的有效性。最后,将蛋白质序列和结构属性适当地纳入基于网络的方法中,可以进一步提高PPI预测的性能,一旦这类信息的规模可以扩大。
本文展现内容并非原文所有内容,具体实验细节可见原文。
-------------------------------------------
欢迎点赞收藏转发!
下次见!