- 迷你世界api 系统事件
yonghumyicunzai
游戏游戏游戏开发
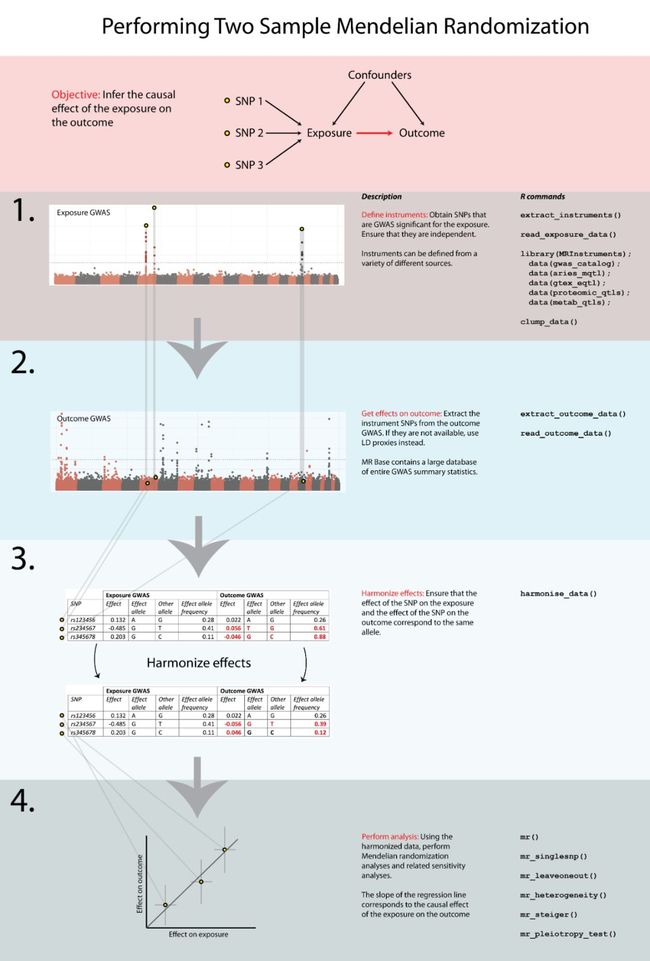
迷你世界api系统事件游戏活动管理只需添加需要监视的事件,而无需创建事件对象,如下所示:--GameEvent---ScriptSupportEvent:registerEvent([=[Game.Start]=],Game_StartGame)ScriptSupportEvent:registerEvent([=[Game.Run]=],Game_Update)ScriptSupportEven
- 申威、龙芯、海光等六大国产芯片前景分析,谁有扛鼎之力?
艾瑞网科技
人工智能
申威、龙芯、海光等六大国产芯片前景分析,谁有扛鼎之力?芯片是底层硬件基础设施的核心,也是智能设备的心脏,人们愈发意识到芯片对于各行各业发展的重要意义,也对国内的芯片厂商投入了更多关注度。经过多年发展,目前我国已有超过14万家芯片相关企业,其中申威、龙芯、海光、兆芯、鲲鹏、飞腾六大厂商作为中坚代表,共同引领着国内芯片产业的进步。这六大厂商谁的商业应用前景更广,谁拥有更可持续发展的未来?其实从现在我们
- 代码随想录算法训练营第七天|Leetcode 344.反转字符串 541. 反转字符串II 卡码网:54.替换数字
昂子的博客
算法leetcodejava数据结构
344.反转字符串建议:本题是字符串基础题目,就是考察reverse函数的实现,同时也明确一下平时刷题什么时候用库函数,什么时候不用库函数题目链接/文章讲解/视频讲解:代码随想录思路非常简单,两个指针一个指向头一个指向尾巴,对于字符串,我们定义两个指针(也可以说是索引下标),一个从字符串前面,一个从字符串后面,两个指针同时向中间移动,并交换元素。classSolution{publicvoidre
- VB6 调用 JS 函数时数据传输json格式或a=1&b=s2字符串
专注VB编程开发20年
javascriptjson开发语言vb6js
1.VB6调用JS函数时数据传输格式当从VB6调用JS设计的函数时,使用JSON字符串作为数据传输格式是一个不错的选择,但并非唯一选择。使用JSON字符串传输的优势通用性:JSON是一种轻量级的数据交换格式,具有良好的跨语言和跨平台特性。在VB6和JS之间使用JSON字符串传输数据,可以方便地表示复杂的数据结构,如对象、数组等。结构化:JSON可以清晰地表示数据的结构,便于在不同语言环境中解析和处
- 如何在 Conda 环境中使用 PySide6 将 .ui 文件转换为 .py 文件
元素之窗
condaui
如何在Conda环境中使用PySide6将.ui文件转换为.py文件在PyQt或PySide6开发中,通常会使用QtDesigner设计UI界面,并生成.ui文件。但为了在Python代码中使用这些UI设计,我们需要将.ui文件转换为.py文件。本文将介绍如何在Conda环境中使用PySide6进行转换。1.确保Conda环境已激活在PowerShell或命令行中,首先激活你的Conda环境,例如
- C++ 泛型编程
四代目 水门
C++学习笔记c++开发语言
C++泛型编程一、泛型编程基础1.核心概念实现算法与数据结构的分离基于模板技术(函数模板/类模板)本质:类型参数化,减少重复代码典型应用:STL容器、迭代器、算法2.类型本质内存布局的抽象不同类型对应不同的内存分配策略二、函数模板1.基本语法cpptemplate//或template返回类型函数名(参数列表){//函数体}2.关键特性支持隐式推导和显式指定类型可重载(包括与普通函数重载)可声明为
- 【leetcode hot 100 54】螺旋矩阵
longii11
leetcode矩阵windows
错误解法:以轮数定义旋转过程进行输出classSolution{publicListspiralOrder(int[][]matrix){Listlist=newLinkedList=round){list.add(matrix[i][j]);j--;}//j++;i--;while(i>=round+1){list.add(matrix[i][j]);i--;}i++;j++;round++;}
- 【学习笔记5】Linux下cuda、cudnn、pytorch版本对应关系
longii11
linuxpytorch运维
一、cuda和cudnnNVIDIACUDAToolkit(CUDA)为创建高性能GPU加速应用程序提供了一个开发环境。借助CUDA工具包,您可以在GPU加速的嵌入式系统、桌面工作站、企业数据中心、基于云的平台和HPC超级计算机上开发、优化和部署您的应用程序。该工具包包括GPU加速库、调试和优化工具、C/C++编译器以及用于部署应用程序的运行时库。全球的深度学习研究人员和框架开发人员都依赖cuDN
- springboot整合rabbitMQ
twx95
java-rabbitmqspringbootrabbitmq
安装rabbitMQ虚拟机或者服务器上安装我这里使用的是vm虚拟机做演示第一步:安装docker参考linux安装docker-CSDN博客第二步:拉取rabbitMQ镜像3-management(镜像版本)dockerpullrabbitmq:3-management查看镜像是否拉取成功dockerimages第三步:运行rabbitMQdockerrun\-eRABBITMQ_DEFAULT_
- python找色_Python获取图片位置像素色值及判断色值是否存在
weixin_39966941
python找色
#!/usr/bin/python#-*-coding:utf-8-*-fromPILimportImage#涛哥用代码看是那的错https://blog.kydbk.comimg=Image.open("test.png")#获取图片尺寸的大小(600,600)printimg.size#获取图片的格式pngprintimg.format#获取图片的图像类型RGBAprintimg.mode#显
- 字节跳动2024校招后端开发面试题大全(含解题思路)
AI天才研究院
ChatGPTAI大模型企业级应用开发实战DeepSeekR1&大数据AI人工智能大模型大厂Offer收割机面试题简历程序员读书硅基计算碳基计算认知计算生物计算深度学习神经网络大数据AIGCAGILLMJavaPython架构设计Agent程序员实现财富自由
字节跳动2024校招后端开发面试题大全(含解题思路)关键词:字节跳动、校招、后端开发、面试题、解题思路摘要:本文将围绕字节跳动2024校招后端开发面试题进行深入分析,包括数据结构与算法、编程语言基础、后端技术栈、微服务架构、系统设计与优化等方面的面试题。通过详细解析这些面试题,帮助读者理解解题思路,提升后端开发面试技能。字节跳动2024校招后端开发面试背景字节跳动(ByteDance)是中国领先的
- react中的useContext--为什么使用(一)
咖啡虫
Reactreact.js前端前端框架
React的数据传递流程在React中,数据传递通常是自上而下的,也就是父组件把数据通过props传递给子组件,子组件无法直接修改父组件的数据。例子:父组件向子组件传递数据constParent=()=>{constuser={name:"John",age:24};return;};constChild=({user})=>{return(姓名:{user.name}年龄:{user.age})
- 读取图片并显示每个像素处的RGB值
vincent-xia
C++GDI像素RGB
不记得在网上什么地方见到的demo了,感觉会有时候会用到。就是读取一幅图片每个像素处的RGB三个通道的值。代码如下,代码很简单,就不解释了,主要是那个gdi中的函数用的少:#include#include#include#include#include#pragmacomment(lib,"gdiplus.lib")usingnamespacestd;usingnamespaceGdiplus;i
- 【Elasticsearch】Index Lifecycle Management
risc123456
Elasticsearchelasticsearch
Elasticsearch的索引生命周期管理(IndexLifecycleManagement,简称ILM)是一种自动化管理索引生命周期的功能,旨在帮助用户根据索引的使用模式和数据价值,高效地管理和优化索引的存储、性能和成本。以下是关于Elasticsearch索引生命周期的详细说明:---1.索引生命周期的五个阶段Elasticsearch的ILM定义了五个主要阶段,每个阶段对应不同的索引使用模
- LeetCode-Hot100-006三数之和
YQ_ZJH
LeetCode100题leetcode数据结构排序算法算法c++蓝桥杯java
思路先排序解决重复的问题。再三重循环遍历,但是第二重和第三重使用双指针的做法,复杂度降低为O(n2)O(n^2)O(n2)。代码本次代码来自于力扣官方题解评论区,非本人原创,请注意classSolution{publicList>threeSum(int[]nums){Arrays.sort(nums);//先排序List>res=newArrayList0&&nums[i]==nums[i-1]
- 【Qt】Qt Widgets和QML(Qt Quick)开发界面的区别
£އއ昔年
qt开发语言
Qt提供了两种主要的UI技术:QtWidgets和QML(QtQuick)。它们的核心区别主要体现在使用方式、架构、性能、开发难度和适用场景等方面。1.QtWidgetsvs.QML总体对比对比项QtWidgetsQML(QtQuick)语言C++(带QtUI库)QML+JavaScript(底层C++)渲染方式传统窗口系统控件(原生或模拟)基于OpenGL,使用GPU加速UI风格经典桌面UI(W
- 将返回的json数据存储为json文件
小凳子在线
json
背景:想下载行政区域数据,网站:unpkg.com/province-city-china@8.5.8/dist/data.json返回json数据(代码由通义生成,此处仅作为记录)工具类://src/utils/downloadData.jsexportasyncfunctiondownloadData(url,filename){try{constresponse=awaitfetch(url
- JAVA排序
荔枝吃吃
java排序算法算法
1.冒泡排序/***使用冒泡排序算法对整数数组进行排序*冒泡排序是一种简单的排序算法,它重复地遍历要排序的数列,*一次比较两个元素,如果它们的顺序错误就把它们交换过来*遍历数列的工作是重复地进行直到没有再需要交换,也就是说该数列已经排序完成*这个算法的名字由来是因为越小(或越大)的元素会经过交换慢慢“浮”到数列的顶端**@paramarr待排序的整数数组*/publicstaticvoidbubb
- 面向B端程序员的逆袭:从码农到业务解决方案架构师的进阶之路
Hello kele
运维人工智能经验分享AI编程程序员
在2B(企业服务)这个深水区,程序员已经不能只当“代码搬运工”了。现在的趋势是从单纯的技术交付,升级成帮客户创造商业价值的“大神”。客户不再只是IT部门那帮geek,连业务部门的老大们也开始掺和进来,解决方案得直击企业数字化转型的痛点。这时候,你的价值就得跟“客户成功”死磕到底。这篇文章从技术穿透力、业务理解度、客户连接力三个角度,给你画一张从码农到业务解决方案架构师的“升级地图”。一、技术穿透力
- 大语言模型对程序员行业的影响及未来发展走势分析
Hello kele
人工智能java人工智能AI编程
随着人工智能技术的快速发展,特别是大语言模型(如DeepSeek、OpenAI、Grok等)的出现,对程序员这个行业产生了深远的影响。在这篇文章中,我们将探讨这些变化,分析影响,并展望未来的发展趋势。一、当前影响1.自动化代码生成大语言模型的一个直接影响是代码自动化的能力。这些模型可以理解代码上下文,并生成功能性代码。例如,GitHubCopilot已经成为许多开发者的辅助工具,能够根据注释或部分
- 【AI辅助工具】Trae和Cursor 对比分析
Hello kele
人工智能AI编程
Trae和Cursor都是旨在提升编程效率的AI辅助工具,但在功能、定位和用户体验上有所差异。Trae:Trae是字节跳动推出的AI集成开发环境(IDE),专为中文开发者设计,提供全中文界面,符合国人使用习惯。主要特点:智能问答与代码自动补全:支持通过自然语言描述需求,自动生成相应的代码,减少手动编写代码的时间。Builder模式:类似于Cursor的Composer功能,帮助用户从零开始构建完整
- DeepSeek:AI赋能的无限可能——从日常生活到职业进阶的全场景探索
Hello kele
人工智能人工智能
引言在人工智能技术飞速发展的今天,DeepSeek作为一款国产AI工具,凭借其强大的推理能力、自然语言处理效率和场景化应用潜力,正在重塑人类解决问题的方式。从撰写演讲稿到制定投资策略,从家庭教育到企业管理,DeepSeek通过“自然语言对话”的交互模式,将复杂任务简化为几步提示词的输入,真正实现了“所想即所得”。本文将从七大核心场景出发,系统解析DeepSeek如何成为个人与组织的智能助手,推动效
- 迷你世界脚本自定义UI接口:Customui
星空露珠
笔记lua游戏数据结构
自定义UI接口:Customui彼得兔更新时间:2024-11-0715:12:42具体函数名及描述如下:(除前两个,其余的目前只能在UI编辑器内部的脚本使用)序号函数名函数描述1openUIView(...)打开一个UI界面(注意:继承自player对象)2hideUIView(...)隐藏一个UI界面(注意:继承自player对象)1setText(...)设置文本元件内容(只在UI局部脚本有
- 迷你世界脚本事件列表:Event
星空露珠
笔记游戏数据结构lua
事件列表:Event彼得兔更新时间:2024-07-2612:04:51直接添加要监听的事件即可,无需自行创建事件管理对象。具体例子如下:--游戏事件---ScriptSupportEvent:registerEvent([=[Game.Start]=],Game_StartGame)ScriptSupportEvent:registerEvent([=[Game.Run]=],Game_Upda
- 迷你世界脚本函数监听接口:ListenParam
星空露珠
笔记游戏数据结构lua
函数监听接口:ListenParam彼得兔更新时间:2023-04-2610:20:18具体函数名及描述如下:序号函数名函数描述1AddGraphicsListenParam(...)添加图文信息监听触发器参数刷新的对象id参数信息回调方法AddGraphicsListenParam参数及类型:graphid:number已创建的图文信息IDfuncs:table监听函数列表param:table
- Leetcode 刷题笔记1 动态规划part05
平乐君
leetcode笔记动态规划
开始完全背包不同于01背包,完全背包的特色在于元素可以重复拿取,因此在递归公式和遍历顺序上都有些许不同。leetcode518零钱兑换||在组合方式中所用到的递推公式是dp[j]=dp[j-coins[i]]+dp[j]对于coins[i]>j的情况,forjinrange(coin[i],amount+1)不会执行,即实现dp[i][j]=dp[i-1][j]classSolution:defc
- Leetcode 刷题笔记1 动态规划part04
平乐君
leetcode笔记动态规划
leetcode最后一块石头的重量||问题转化,把石头问题转化为背包问题,在target容量范围内所能装的最大石头重量classSolution:deflastStoneWeightII(self,stones:List[int])->int:total=sum(stones)target=total//2dp=[0]*(target+1)forstoneinstones:forjinrange(
- IMT-2020(5G)推进组发布《5G-Advanced 场景需求与关键技术白皮书》
优橙教育
5G面试职场和发展5g网络
11月16日,由工业和信息化部、深圳市人民政府主办的2022年中国5G发展大会在深圳举行。本届大会以“5G领航新基建,构筑发展新底座”为主题。会上,IMT-2020(5G)推进组发布《5G-Advanced场景需求与关键技术白皮书》。中国工程院院士邬贺铨表示,5G商用三年来在国际上取得了网络部署与用户数领先的成绩。2022年9月中国建成5G基站数占基站总数的20.6%,占全球5G基站数60%。20
- [数据结构] [C++ STL] vector使用详解
高亚奇
数据结构数据结构c++开发语言
一、概述vector(向量):是一种序列式容器,事实上和数组差不多,但它比数组更优越。一般来说数组不能动态拓展,因此在程序运行的时候不是浪费内存,就是造成越界。而vector正好弥补了这个缺陷,它的特征是相当于可分配拓展的数组(动态数组),它的随机访问快,在中间插入和删除慢,但在末端插入和删除快。二、定义及初始化使用之前必须加相应容器的头文件:#include//vector属于std命名域的,因
- 前端React篇之哪些方法会触发 React 重新渲染?重新渲染 render 会做些什么?
m0_74823705
前端react.jsjavascript
目录哪些方法会触发React重新渲染?重新渲染render会做些什么?setState()案例需求总结forceUpdate()案例需求总结props改变案例需求总结context改变案例需求总结哪些方法会触发React重新渲染?重新渲染render会做些什么?在React中,以下方法会触发重新渲染:setState():当调用组件的setState方法并传入新的状态值时,React会触发重新渲染
- 矩阵求逆(JAVA)利用伴随矩阵
qiuwanchi
利用伴随矩阵求逆矩阵
package gaodai.matrix;
import gaodai.determinant.DeterminantCalculation;
import java.util.ArrayList;
import java.util.List;
import java.util.Scanner;
/**
* 矩阵求逆(利用伴随矩阵)
* @author 邱万迟
- 单例(Singleton)模式
aoyouzi
单例模式Singleton
3.1 概述 如果要保证系统里一个类最多只能存在一个实例时,我们就需要单例模式。这种情况在我们应用中经常碰到,例如缓存池,数据库连接池,线程池,一些应用服务实例等。在多线程环境中,为了保证实例的唯一性其实并不简单,这章将和读者一起探讨如何实现单例模式。 3.2
- [开源与自主研发]就算可以轻易获得外部技术支持,自己也必须研发
comsci
开源
现在国内有大量的信息技术产品,都是通过盗版,免费下载,开源,附送等方式从国外的开发者那里获得的。。。。。。
虽然这种情况带来了国内信息产业的短暂繁荣,也促进了电子商务和互联网产业的快速发展,但是实际上,我们应该清醒的看到,这些产业的核心力量是被国外的
- 页面有两个frame,怎样点击一个的链接改变另一个的内容
Array_06
UIXHTML
<a src="地址" targets="这里写你要操作的Frame的名字" />搜索
然后你点击连接以后你的新页面就会显示在你设置的Frame名字的框那里
targerts="",就是你要填写目标的显示页面位置
=====================
例如:
<frame src=&
- Struts2实现单个/多个文件上传和下载
oloz
文件上传struts
struts2单文件上传:
步骤01:jsp页面
<!--在进行文件上传时,表单提交方式一定要是post的方式,因为文件上传时二进制文件可能会很大,还有就是enctype属性,这个属性一定要写成multipart/form-data,不然就会以二进制文本上传到服务器端-->
<form action="fileUplo
- 推荐10个在线logo设计网站
362217990
logo
在线设计Logo网站。
1、http://flickr.nosv.org(这个太简单)
2、http://www.logomaker.com/?source=1.5770.1
3、http://www.simwebsol.com/ImageTool
4、http://www.logogenerator.com/logo.php?nal=1&tpl_catlist[]=2
5、ht
- jsp上传文件
香水浓
jspfileupload
1. jsp上传
Notice:
1. form表单 method 属性必须设置为 POST 方法 ,不能使用 GET 方法
2. form表单 enctype 属性需要设置为 multipart/form-data
3. form表单 action 属性需要设置为提交到后台处理文件上传的jsp文件地址或者servlet地址。例如 uploadFile.jsp 程序文件用来处理上传的文
- 我的架构经验系列文章 - 前端架构
agevs
JavaScriptWeb框架UIjQuer
框架层面:近几年前端发展很快,前端之所以叫前端因为前端是已经可以独立成为一种职业了,js也不再是十年前的玩具了,以前富客户端RIA的应用可能会用flash/flex或是silverlight,现在可以使用js来完成大部分的功能,因此js作为一门前端的支撑语言也不仅仅是进行的简单的编码,越来越多框架性的东西出现了。越来越多的开发模式转变为后端只是吐json的数据源,而前端做所有UI的事情。MVCMV
- android ksoap2 中把XML(DataSet) 当做参数传递
aijuans
android
我的android app中需要发送webservice ,于是我使用了 ksop2 进行发送,在测试过程中不是很顺利,不能正常工作.我的web service 请求格式如下
[html]
view plain
copy
<Envelope xmlns="http://schemas.
- 使用Spring进行统一日志管理 + 统一异常管理
baalwolf
spring
统一日志和异常管理配置好后,SSH项目中,代码以往散落的log.info() 和 try..catch..finally 再也不见踪影!
统一日志异常实现类:
[java]
view plain
copy
package com.pilelot.web.util;
impor
- Android SDK 国内镜像
BigBird2012
android sdk
一、镜像地址:
1、东软信息学院的 Android SDK 镜像,比配置代理下载快多了。
配置地址, http://mirrors.neusoft.edu.cn/configurations.we#android
2、北京化工大学的:
IPV4:ubuntu.buct.edu.cn
IPV4:ubuntu.buct.cn
IPV6:ubuntu.buct6.edu.cn
- HTML无害化和Sanitize模块
bijian1013
JavaScriptAngularJSLinkySanitize
一.ng-bind-html、ng-bind-html-unsafe
AngularJS非常注重安全方面的问题,它会尽一切可能把大多数攻击手段最小化。其中一个攻击手段是向你的web页面里注入不安全的HTML,然后利用它触发跨站攻击或者注入攻击。
考虑这样一个例子,假设我们有一个变量存
- [Maven学习笔记二]Maven命令
bit1129
maven
mvn compile
compile编译命令将src/main/java和src/main/resources中的代码和配置文件编译到target/classes中,不会对src/test/java中的测试类进行编译
MVN编译使用
maven-resources-plugin:2.6:resources
maven-compiler-plugin:2.5.1:compile
&nbs
- 【Java命令二】jhat
bit1129
Java命令
jhat用于分析使用jmap dump的文件,,可以将堆中的对象以html的形式显示出来,包括对象的数量,大小等等,并支持对象查询语言。 jhat默认开启监听端口7000的HTTP服务,jhat是Java Heap Analysis Tool的缩写
1. 用法:
[hadoop@hadoop bin]$ jhat -help
Usage: jhat [-stack <bool&g
- JBoss 5.1.0 GA:Error installing to Instantiated: name=AttachmentStore state=Desc
ronin47
进到类似目录 server/default/conf/bootstrap,打开文件 profile.xml找到: Xml代码<bean
name="AttachmentStore"
class="org.jboss.system.server.profileservice.repository.AbstractAtta
- 写给初学者的6条网页设计安全配色指南
brotherlamp
UIui自学ui视频ui教程ui资料
网页设计中最基本的原则之一是,不管你花多长时间创造一个华丽的设计,其最终的角色都是这场秀中真正的明星——内容的衬托
我仍然清楚地记得我最早的一次美术课,那时我还是一个小小的、对凡事都充满渴望的孩子,我摆放出一大堆漂亮的彩色颜料。我仍然记得当我第一次看到原色与另一种颜色混合变成第二种颜色时的那种兴奋,并且我想,既然两种颜色能创造出一种全新的美丽色彩,那所有颜色
- 有一个数组,每次从中间随机取一个,然后放回去,当所有的元素都被取过,返回总共的取的次数。写一个函数实现。复杂度是什么。
bylijinnan
java算法面试
import java.util.Random;
import java.util.Set;
import java.util.TreeSet;
/**
* http://weibo.com/1915548291/z7HtOF4sx
* #面试题#有一个数组,每次从中间随机取一个,然后放回去,当所有的元素都被取过,返回总共的取的次数。
* 写一个函数实现。复杂度是什么
- struts2获得request、session、application方式
chiangfai
application
1、与Servlet API解耦的访问方式。
a.Struts2对HttpServletRequest、HttpSession、ServletContext进行了封装,构造了三个Map对象来替代这三种对象要获取这三个Map对象,使用ActionContext类。
----->
package pro.action;
import java.util.Map;
imp
- 改变python的默认语言设置
chenchao051
python
import sys
sys.getdefaultencoding()
可以测试出默认语言,要改变的话,需要在python lib的site-packages文件夹下新建:
sitecustomize.py, 这个文件比较特殊,会在python启动时来加载,所以就可以在里面写上:
import sys
sys.setdefaultencoding('utf-8')
&n
- mysql导入数据load data infile用法
daizj
mysql导入数据
我们常常导入数据!mysql有一个高效导入方法,那就是load data infile 下面来看案例说明
基本语法:
load data [low_priority] [local] infile 'file_name txt' [replace | ignore]
into table tbl_name
[fields
[terminated by't']
[OPTI
- phpexcel导入excel表到数据库简单入门示例
dcj3sjt126com
PHPExcel
跟导出相对应的,同一个数据表,也是将phpexcel类放在class目录下,将Excel表格中的内容读取出来放到数据库中
<?php
error_reporting(E_ALL);
set_time_limit(0);
?>
<html>
<head>
<meta http-equiv="Content-Type"
- 22岁到72岁的男人对女人的要求
dcj3sjt126com
22岁男人对女人的要求是:一,美丽,二,性感,三,有份具品味的职业,四,极有耐性,善解人意,五,该聪明的时候聪明,六,作小鸟依人状时尽量自然,七,怎样穿都好看,八,懂得适当地撒娇,九,虽作惊喜反应,但看起来自然,十,上了床就是个无条件荡妇。 32岁的男人对女人的要求,略作修定,是:一,入得厨房,进得睡房,二,不必服侍皇太后,三,不介意浪漫蜡烛配盒饭,四,听多过说,五,不再傻笑,六,懂得独
- Spring和HIbernate对DDM设计的支持
e200702084
DAO设计模式springHibernate领域模型
A:数据访问对象
DAO和资源库在领域驱动设计中都很重要。DAO是关系型数据库和应用之间的契约。它封装了Web应用中的数据库CRUD操作细节。另一方面,资源库是一个独立的抽象,它与DAO进行交互,并提供到领域模型的“业务接口”。
资源库使用领域的通用语言,处理所有必要的DAO,并使用领域理解的语言提供对领域模型的数据访问服务。
- NoSql 数据库的特性比较
geeksun
NoSQL
Redis 是一个开源的使用ANSI C语言编写、支持网络、可基于内存亦可持久化的日志型、Key-Value数据库,并提供多种语言的API。目前由VMware主持开发工作。
1. 数据模型
作为Key-value型数据库,Redis也提供了键(Key)和值(Value)的映射关系。除了常规的数值或字符串,Redis的键值还可以是以下形式之一:
Lists (列表)
Sets
- 使用 Nginx Upload Module 实现上传文件功能
hongtoushizi
nginx
转载自: http://www.tuicool.com/wx/aUrAzm
普通网站在实现文件上传功能的时候,一般是使用Python,Java等后端程序实现,比较麻烦。Nginx有一个Upload模块,可以非常简单的实现文件上传功能。此模块的原理是先把用户上传的文件保存到临时文件,然后在交由后台页面处理,并且把文件的原名,上传后的名称,文件类型,文件大小set到页面。下
- spring-boot-web-ui及thymeleaf基本使用
jishiweili
springthymeleaf
视图控制层代码demo如下:
@Controller
@RequestMapping("/")
public class MessageController {
private final MessageRepository messageRepository;
@Autowired
public MessageController(Mes
- 数据源架构模式之活动记录
home198979
PHP架构活动记录数据映射
hello!架构
一、概念
活动记录(Active Record):一个对象,它包装数据库表或视图中某一行,封装数据库访问,并在这些数据上增加了领域逻辑。
对象既有数据又有行为。活动记录使用直截了当的方法,把数据访问逻辑置于领域对象中。
二、实现简单活动记录
活动记录在php许多框架中都有应用,如cakephp。
<?php
/**
* 行数据入口类
*
- Linux Shell脚本之自动修改IP
pda158
linuxcentosDebian脚本
作为一名
Linux SA,日常运维中很多地方都会用到脚本,而服务器的ip一般采用静态ip或者MAC绑定,当然后者比较操作起来相对繁琐,而前者我们可以设置主机名、ip信息、网关等配置。修改成特定的主机名在维护和管理方面也比较方便。如下脚本用途为:修改ip和主机名等相关信息,可以根据实际需求修改,举一反三!
#!/bin/sh
#auto Change ip netmask ga
- 开发环境搭建
独浮云
eclipsejdktomcat
最近在开发过程中,经常出现MyEclipse内存溢出等错误,需要重启的情况,好麻烦。对于一般的JAVA+TOMCAT项目开发,其实没有必要使用重量级的MyEclipse,使用eclipse就足够了。尤其是开发机器硬件配置一般的人。
&n
- 操作日期和时间的工具类
vipbooks
工具类
大家好啊,好久没有来这里发文章了,今天来逛逛,分享一篇刚写不久的操作日期和时间的工具类,希望对大家有所帮助。
/*
* @(#)DataFormatUtils.java 2010-10-10
*
* Copyright 2010 BianJing,All rights reserved.
*/
package test;
impor