宏转录组方法_Cell:基因表达的改变和群落的更替塑造了全球海洋宏转录组
编译:jiee,编辑:小菌菌、江舜尧。
导读
海洋微生物群落极大地影响着地球的生物地球化学循环、食物网和气候。尽管最近在理解海洋微生物的物种和基因组组成方面取得了进展,但对其转录组在全球范围内的变化知之甚少。在此研究中,作者呈现了来自全球126个采样站的187个宏转录组和370个宏基因组的综合数据集,并构建了一个包含4700万个基因的参考基因集,来研究全球海洋不同深度层的微生物群落的转录组。作者研究了塑造海洋微生物转录组的基因表达变化和群落更替随着环境变化的潜在机制,并展示了它们在多个生物地球化学相关过程中的个体贡献的不同。此外,作者还发现在极地水域中,基因表达变化的相对贡献明显低于非极地水域,并提出假设:极地地区微生物活动的变化会响应海洋变暖,且生物组成的变化对微生物活动的变化的驱动作用比基因调控机制的驱动作用更强。
论文ID
原名:Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome
译名:基因表达的改变和群落的更替塑造了全球海洋宏转录组
期刊:Cell
IF:36.216
发表时间:2019.11
通讯作者:Shinichi Sunagawa
作者单位:苏黎世联邦理工学院微生物研究所和瑞士生物信息学研究所
实验设计
利用TARA ocean项目在全球126个采样点上采集了187个宏转录组和370个宏基因组样本,进行深度宏基因组测序。综合宏基因组和宏转录组数据得到了最全面的海洋微生物参考基因集(OM-RGC.v2)。然后利用宏基因组和宏转录组分别得到了分类组成、宏基因组组成以及宏转录组组成数据。对得到的宏基因组和宏转录组组成进行归一化和转化,并进行基因表达谱的计算。然后比较了微生物地球化学循环标记基因在深度和纬度上的差异表达和丰度变化。对潜在的固氮微生物进行了宏基因组组装基因组(MAGs)水平上的挖掘。画图数据和代码共享于github:https://github.com/SushiLab/omrgc_v2_scripts。
结果
1.全球海洋微生物研究的新多组学资源
本研究的数据集包括从全球126个采样点上采集的187个宏转录组和370个宏基因组样本,跨越142°的纬度范围(图1;https://doi.org/10.5281/zenodo.3473199)。样本来源于透光层,表层海水(SRF),叶绿素浓度最大值(DCM),混合水层,及中深海的黑暗水层(MES),深度从5米到1000米(SRF, DCM, MES层的平均深度分别为5米,50米和550米)。在对低RNA样本进行方案优化(见STAR方法)后,得到了187个原核生物富集的宏转录组文库,每个样本的平均测序深度为28 G(https://doi.org/10.5281/zenodo.3473199)。这些数据与一组包含131个病毒富集的、59个巨型病毒富集的和180个原核生物富集的宏基因组数据集一起分析(https://doi.org/10.5281/zenodo.3473199),这组数据集中包括以前测序的Tara Oceans数据、44个极地和42个非极地区的病毒富集的宏基因组(见STAR方法),以及41个从北冰洋获取的原核生物富集的宏基因组(本研究中新获得)。
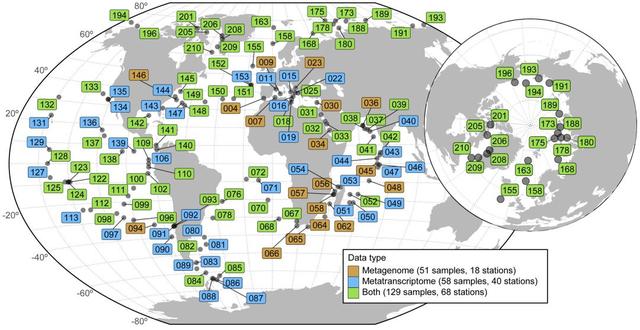
图1 本研究分析的宏组学数据的地理分布情况。Tara Oceans采样站的地理分布(2009 - 2013)。每个站点从不同深度层收集了不同大小的样本,共557个样本(370个宏基因组和187个宏转录组)。编号为155及以上的监测站代表了2013年6月至10月期间开展的TaraOceans极地圈采样站点。颜色表示在每个工作站收集的原核生物富集部分样本的类型:18个工作站只有宏基因组(橙色);40个工作站仅有宏转录组(蓝色);68个工作站至少一个深度层同时有宏基因组和宏转录组。
作者的目标是获得整个微生物群落层面的群落更替和基因表达变化,并将这些数据放在全球范围的地理和环境梯度的背景下。值得注意的是,这种方法的适用与否很大程度上取决于环境中存在的生物体与基因组序列数据库中代表性生物体之间的进化距离。理想情况下,组成相关群落的所有生物体的基因组序列都已被测序,从而促进基因丰度和基因表达数据的整合,以评估整个群落的组成。这样的分析似乎可以用于人类肠道微生物组,因为最近已经获得了理想的肠道微生物基因组资源。然而,对于海洋微生物组样本,在物种水平下,利用目前所有的基因组序列数据库仅能注释到低于10%(宏转录组中)和不到5%(宏基因组中)的微生物(图2A)。

图2 OM-RGC.v2基因的检测率和注释。(A)180个原核生物富集的宏基因组(橙色)和187个原核生物富集的宏转录组(蓝色)中reads的注释比例。(B)180个原核细胞富集的样品中OM-RGC.v2基因集的累积。虚线将139个非北极宏基因组与41个北极宏基因组分开。坡度的增加反映了北冰洋中新基因检测率的增加。(C)基因在域水平上的物种注释(包括病毒,LCA:最近共同祖先),并将基因的功能注释分为9000个KEGG和76000个eggNOG同源基因群(简写为KOs和OGs)。未注释部分的基因用于产生新的基因簇(GCs),以进一步描述基因集的功能。
为了克服这一限制,作者基于地理分布更广的370个宏基因组生成了更新版的海洋微生物参考基因集(OM-RGC.v2),特别包含了北冰洋地区(图1)。在4700万个非冗余基因中,24.5%是从北冰洋样品中重建的,虽然部分基因可以在其他地方检测到(图2),突出了在未开发环境中取样的价值。利用这一参考基因集,近70%的基因可以被分类注释,61%的基因与已知的OG基因具有同源性(eggNOG version 4.5)(见STAR方法)。作者进一步将OM-RGCv2中剩余的39%的未注释基因根据序列相似性进行了聚类,得到250000个基因簇(GCs)(图2C;见STAR方法)。作者在比较不同深度层之间、极地和非极地之间、以及与环境参数相关的基因的转录丰度时,分别找到了5439个GCs、31339个GCs及21648个GCs的显著差异表达(图S2)。这些发现暗示了这些基因与环境变化具有生态相关性但是功能未知。作为一种识别功能相关基因的方法,保守共表达基因分析表明,这些未知的GCs可能参与了信号转导、转录调控和能量产生/转换(图S3;表S1)。与现有海洋基因组参考数据库相比,作者发现本研究中的OM-RGC v2包含了宏基因组和宏转录组中的大部分编码基因(分别为70%和51%)(图2),使其适合分析整个群体的宏转录组组成。所有基因序列的丰度、表达和地理分布都可以在线查询,并且它们与环境参数相互关联,这有助于在未来进行更多的以基因为中心的研究。
2.跨纬度和深度的宏组学组成变化
建立了用来量化整个群落的分类、基因组和转录组成的资源后,作者接下来试图在全球范围内识别海洋生物群落随主要环境的变化模式和驱动因素。大量研究表明,海洋中的微生物群落是垂直分层的,在表明光合层和中深层之间有一个明显的分界线。极地和非极地群落也被证明具有不同的物种水平的分类学组成。然而,关键的是,同一物种的不同菌株之间共同的基因可能低至40%,例如在大肠杆菌中。此外,微生物群落中的基因功能冗余(相同功能的基因被不同的物种编码)可能有助于在生物多样性丧失的情况下维持重要的群落功能。因此,很难预测基因功能组成和基因表达调控的转录组是否会遵循相同的物种组成变化模式。为了解决这个问题,作者首先去定位了沿纬度梯度分布的表层光合层水域(SRF和DCM)样品(原核生物富集的宏基因组和宏转录组样品)的分化边界。从赤道向北,直到北纬40°才发现明显的表层光合层水域分化。在这个分化点,所有分析中群落组成的分化程度都显著增加,在北纬60°时达到分化峰值。在南半球也观察到了类似的趋势(图3),符合细菌物种组成在极地和非极地水域间的差别。作者进一步发现这种向高纬度的分化反映在一些显著富集的物种(OTUs)中:Flavobacteriales目(如Formosa, Polaribacter, NS5, NS7,和NS9海洋类群),Gammaproteobacteria 纲(OM182类群和Piscirickettsiaceae),真核生物(如Phaeocystis),及Rhodospirillaceae科的Prochlorococcus spp,SAR11和SAR406类群中的部分物种(图S4)。在这里,宏基因组和宏转录组在OGs水平上基因相对丰度和转录拷贝变化的一致性表明:在全球范围内,物种组成在很大程度上决定了功能基因的组成。在塑造跨越生态边界的群落水平的转录组成分方面,物种组成也决定着基因调控变异。

图3 全球海洋微生物群落组成的纬度分区。左边的示意图说明了生态分化的裂动窗分析的基本概念。它包括假定边界两边的群落之间的两两距离与同一边群落之间的两两距离的比较。高微分值表示边界两边的距离比两边内的距离大。窗口宽度分析的间隔为10个样本,显示了原核生物富集的样本(SRF)和深叶绿素最大值水样(DCM)的以北纬60°为中心的基于分类组成(灰色,OTU的相对丰度)、宏基因组组成(橙色,每个基因的丰度),和宏转录组组成(蓝色,每个转录本的相对丰度)的生态边界。类似的模式在南半球也很明显;然而,对生态边界的检测受限于样品数量。显著性是用纬度值的10,000个随机排列计算的99%置信区间来确定的。垂线代表了显著值的纬度范围的窗口。由于样本数量和纬向覆盖范围的限制,无法对中深层进行分析。
事实上,作者发现不同分析方法得到的群落组成都是高度相关的(图S5),且它们在海洋表层的变化可以用27个环境参数中的海水温度最好地解释(图4A)。这个结果补充了早些时候:温度作为一个重要的因素推动海洋微生物群落的分类组成的报道,此后分析的在具有地理效应的全球样本间,温度区别于其他环境参数被证实了在非极地开放海域作为一个关键的因素驱动了物种和基因功能在光合层的组成。事实上,本研究中确定的从北纬40度开始到北纬60度达到顶峰的生态界线,与取样的北大西洋和北极水域之间的温度急剧下降相一致(图S6),且与其他海洋学特征有关。在北纬/南纬40°时,年平均14°等温线有效地将永久分层的海洋与亚极地和极地区域分开,而在北纬60°时北大西洋冬季混合最强烈(混合层深度最深)。因此,作者在这里所描述的微生物群落组成的生态边界可能是由于海洋水团垂直混合变化所引起的物理化学变化,这种变化与海洋表面温度的差异有关。
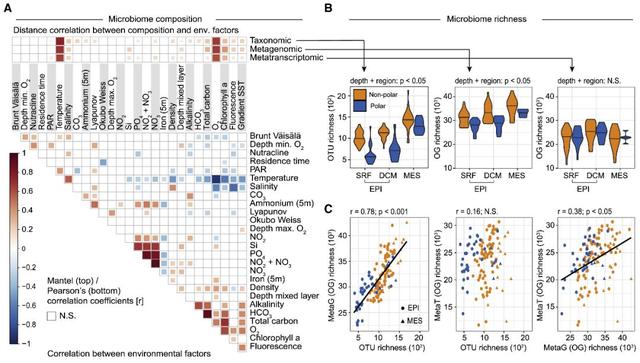
图4 全球海洋微生物群落组成的模式和驱动因素。(A)光合层样品中分类、宏基因组和宏转录组物种组成与27个环境因子的相关性。环境因素的两两比较如下所示,颜色梯度表示spearman相关系数。温度是光合层所有剖面的最佳解释变量,其次是氧浓度,氧浓度与温度高度相关。(B)极地和非极地三个深度层样品中微生物群落的组成丰富度。分类学和功能宏基因组丰富度(OTUs和OGs的数量)随深度增加而增加,丰富度在极地样本中始终低于非极地样本。相比之下,无论是跨深度还是极地和非极地之间的功能基因的丰富度(OGs数量)均无显著差异。小提琴图表示数据的(镜像)密度分布,中位数值显示为一条水平线。(C)物种丰富度(OTUs数量)、功能宏基因组(metaG)丰富度和宏转录组 (metaT)丰富度(OGs数量)之间的相关性。在进行丰富度计算之前,对数据进行均一化处理。所有比较均采用Pearson相关分析。实线对应于最佳线性拟合。
接下来,作者将宏转录组的多度(通过cDNA测序检测到的特异OGs的数量)量化,作为转录基因功能多样性的代表,并将其与物种和宏基因组多度(DNA测序中分别检测到的OTUs和OGs的数量)进行比较。作为多样性的衡量标准,后两者提供了关于生态群落稳定性、功能性及可能的生产力的信息。此外,作者还试图通过比较宏转录组和宏基因组的多度,来量化特定时间下特定群落中编码基因功能实际转录的比例。
物种分类和宏基因组多度高度相关,且没有饱和的迹象,支持先前的研究:海洋生态系统功能冗余相当低;且在所有采样的深度里,物种分类和宏基因组多度在极地都显著低于非极地(图4B)。这些数据与之前的研究一致,表明物种多样性随着纬度的升高而降低,且基因功能多样性同样降低;但是也有其他研究者提出了替代的纬度多样性梯度模型。而宏转录组的多度与物种多度不相关,与宏基因组多度的相关性较差,且极性与非极性地区的菌群之间、各深度层之间差异均不显著(图4B)。这种出乎意料的宏基因组和宏转录组间的多度差异表明,特定宏基因组中的非转录比例在中深层水域和非极地地区要高于光合层水域和极地地区。这可能是由于中深层微生物休眠、死亡以及被动下沉的比例高于光合层。或者,这些观察可能反映了在表层海水中基因组精简的流行,表层海水中的基因组中的基因数量更少。因此,转录基因的比例高于中深层水域。尽管编码功能基因的数量在增加,但同时转录的基因明显饱和,还需要进一步的研究来确定这在其他生物群落中是否也是常见的特征。
3.生物地球化学循环基因的丰度和表达差异
微生物群落转录本库可能会随着环境梯度的变化而变化,作为群落更替和/或基因表达变化的功能(图S1和S7;STAR方法)。为了解开这些参与相关生态过程的基因的个体贡献及它们随环境变化的机制,作者整合了122个原核生物富集的、相互匹配的宏转录组和宏基因组,定量分析了一组生物地球化学标记基因在极地和非极地不同深度水体中的差异丰度和表达水平(图5)。
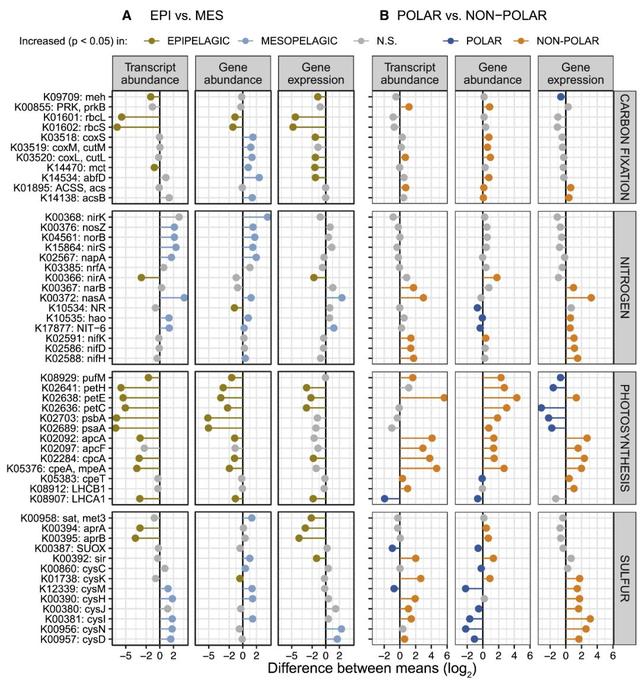
图5 基因丰度和表达的差异决定了代谢标记基因在极地和非极地区不同深度层的转录丰度差异。(A和B)基因和转录的丰度差异,以及代谢标记基因(KOs)在光合层和中深层之间的基因表达水平差异(A),在极地和非极地之间(B)。
首先,作者试图验证数据质量和分析方法,测试在研究较为透彻的过程(包括碳固定、光合作用和氮循环)中是否可以观察到基因模式。正如所料,作者发现,光合层和中深层之间丰度最不相同的转录本包括光合作用标记基因psaA和psbA以及碳固定中的关键酶-编码RuBisCO的亚基(rbcL和rbcS)(图5A)。此外,作者发现rbcL和rbcS的丰度与psaA和psbB高度相关,这与预期一致:说明固碳作用主要是由光合自养微生物驱动的而不是化能自养菌。尽管有化学自养生物存在,但在中深层水域中RuBisCO基因表达水平较低,进一步证实了上述观点(图S8)。除psbA,其他光合标记基因,包括光合反应中心(petC、petE、petH)和蓝藻特异天线蛋白(apcA、apcF、cpcA、cpeA、cpeT),在极地都比非极地丰度低(图5B)。这一结果可能反映了较冷环境中蓝藻细菌的减少(图S4),或者作者在此分析的原核生物富集的样品中缺乏真核光养生物。
关于氮循环,作者检测了反硝化过程中的标记基因(napA、nirS、norB和nosZ)在光合层和中深层水域中富集的基因和转录本丰度(图5A)。正如预期的那样,这个主要的厌氧过程中的转录本丰度在缺氧的水域中特别高,但是在一些氧气充足的北极水样中也观察到类似的转录水平(图S9)。固氮标记基因(nifK、nifH和nifD)在非极地比在极地更丰富,在20~35(绝对纬度)的水域中含量最高,该水域中硝酸盐和亚硝酸盐浓度低(图S10)。这些数据普遍符合长期以来的认知,即在氮限制条件下固氮活性更高,主要受热带和亚热带地区的蓝藻菌驱动。然而,最近的研究提供了额外的证据支持:在更大的地理和深度范围中具有更广泛的非蓝细菌的异养的固氮生物。鉴于这些发现,作者进一步深入研究了nifH基因的生物地理分布,并确定了哪些生物不仅编码该基因,而且具有表达活性。具体来说,作者分析了122个相互匹配的宏基因组和宏转录组中检测到的24个编码nifH物种的基因分布和转录丰度。从这一分析中,我们发现了一类Gamma-和Deltaproteobacteria纲的物种,它们的基因组最近才被重建,它们不仅数量丰富,而且是本研究样本中nifH转录池的主要贡献者(图6)。此外,作者第一次在中深层的北极水域中发现nifH基因的表达,并重建了携带了该nif操纵子结构物种的基因组(http://doi.org/10.5281/zenodo.3352180,见STAR方法)。该物种可能属于异养的Deltaproteobacterium纲或是Myxococcota门(最近提议一个标准化的细菌分类方法),这需要进一步鉴定。
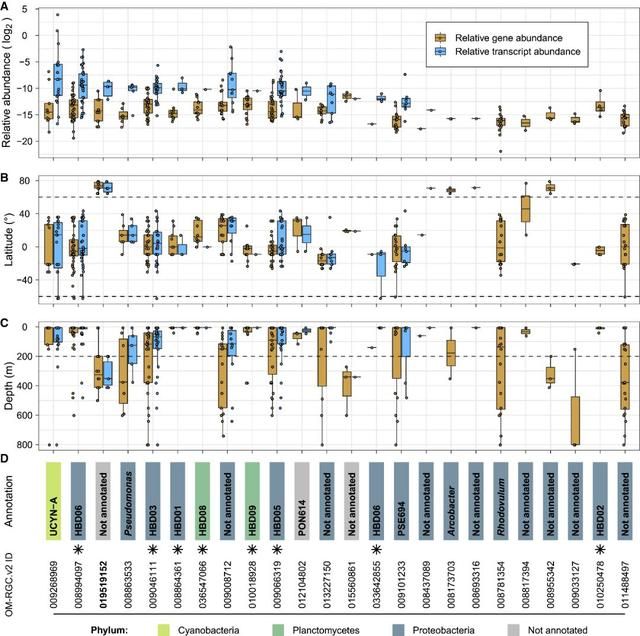
图6. 24个编码nifH “物种”的相对基因和转录丰度。(A)OM-RGC.v2中注释到的24个nifH基因在122个相应的的宏基因组和宏转录组中的相对基因(橘黄色)和转录(淡蓝色)丰度分布;(B)按纬度显示和分类;(C)按深度显示和分类。(D)颜色表示门水平的物种注释,命名对应于更精细的分类或数据库特异性标识符,星号表示之前在异养细菌重氮营养体(HBDs)的MAGs中识别到的基因。(B)和(C)中的水平虚线表示:用以定义极地和非极地的纬度及用以区分光合层和中深层的深度。
尽管作者在超过3年的时间内收集的空间离散数据以及采样过程中存在着固有偏差(例如在取样过程中,转录丰度的季节性或潜在变化的影响无法解释),但是作者能够在全球范围内利用宏转录组数据验证预期的代谢模式。除了验证方法外,作者还演示了如何将以群落为中心的宏转录组分析方法与宏基因组数据结合使用,并进一步为新的基因组解析搭建桥梁。基于分析的稳健性,作者接下来揭示了不同深度和纬度的群落转录组差异的机制。值得注意的是,在作者观察到的案例中,转录丰度的变化可能主要归因于基因丰度或基因表达的差异,也可能是这些机制的组合。如上所述,光合层水域和中深层水域中反硝化标记基因转录本的富集主要是由基因丰度的变化驱动的(图5A)。在这种情况下,中深层水域较高的硝酸盐和亚硝酸盐浓度对生物群落组成的环境过滤导致了基因丰度的变化,从而主导了所观察到的群落转录组差异。相反,由这些基因表达增加所驱动的厌氧异化硫酸盐还原标记基因(aprA和aprB)在光合层水域中转录丰度较高,尽管这些基因的丰度在各深度层之间没有显著差异(图5A)。分类分析表明,有39%和59%的aprA和aprB基因是由Proteobacteria编码的,每个基因中只有2%的基因属于已知的能够进行硫酸盐还原的细菌类群(Archaea, Firmicutes, Nitrospirae, 和Deltaproteobacteria)。这些结果表明aprA和aprB在氧化的水域中替代用途的意义,即通过氧化在细胞质中累积的亚硫酸盐来为细胞解毒,正如SAR11和SAR116中的情况一样且可能在全球均有分布。
在转录组中观察大量同化硫酸盐还原标记基因(cysD, cysH, cysI、cysJ cysN)的差异更为复杂,群落更替和基因表达变化共同导致这些基因在纬度梯度的差异变化(即非极地区比极地地区有更高转录丰度)。在这种情况下,尽管基因的丰度较低,但非极地水域中转录丰度的增加是由于更高的表达水平。有趣的是,作者发现这些标记基因的转录丰度与dmdA负相关(图S11),dmdA是DMSP脱甲基的关键基因,而DMSP可以将碳和硫结合到细菌生物量中。基于这些数据,同时DMSP被原核生物用作硫同化的替代来源,作者提出假设同化性硫酸盐还原途径的全球表达可能随可获取DMSP含量的下降而下调。值得注意的是,如果周转和差异基因表达都起作用的话,仅依靠基因丰度可能会得到错误的、与转录水平相反的预测(例,非光合碳途径中的mct和abfD在光合层中具有较高的表达水平,但在中深层中基因丰度较高)。
4.极性水体群落的转变导致了基因表达的差异
在全球气候变化的背景下,迫切需要更好地了解海洋微生物群落将如何应对正在发生的气候变化。特别是,北极地区经历了一些迄今为止有记录的最高的海洋表层水温变化。海洋变暖模型预测:本世纪末北极地区的平均地表水温将增加2至5摄氏度,突出了更好地理解这些变化对微生物群落的影响的迫切性。鉴于这些预测集中于地表温度变化以及由于它们的变化对生物地球化学循环的主要影响,作者试图评估这些由宏转录组反应出的表层微生物对环境变化的响应,然后利用这些空间离散的数据对未来的预测进行假设。
具体地说,作者的目的是阐明采样点中的微生物群落转录组的差异随着温度梯度的变化是否更强烈地受到群落更替和/或基因表达的影响(图S7; STAR方法)。为此,作者使用一个沿着温度梯度的滑动窗口,将所有样本分成15个样本组,这样每个组都反映了本世纪末海洋变暖的预期温度范围(每个组内温度中位值为1.6°;图S12A)。然后,作者量化了每个组中宏转录组变化的不同机制(图7;STAR方法),发现在温暖的光合层水域中,群落更替对宏转录组组成差异的相对贡献明显低于基因表达变化。相反,在较冷(主要是北极)水域,群落更替的影响高于或与基因表达变化的影响力相同(图7A)。总体而言,极地群落的更替显著高于非极地群落(p<0.001),而基因表达变化呈现相反的模式(p<0.001)(图7B)。有趣的是,不同的宏转录组变化机制的相对贡献在15°时发生了转变,这与之前确定的生态边界相吻合,因此,这不仅描述了不同组成的群落,而且也描述了它们形成不同转录组的机制。作者进一步发现,温度的影响大于其他环境变量(如硝酸盐/亚硝酸盐浓度和盐度)(图S12),这表明在温暖的海洋中,微生物群落对温度变化的响应能力高于寒冷的光合层水域。
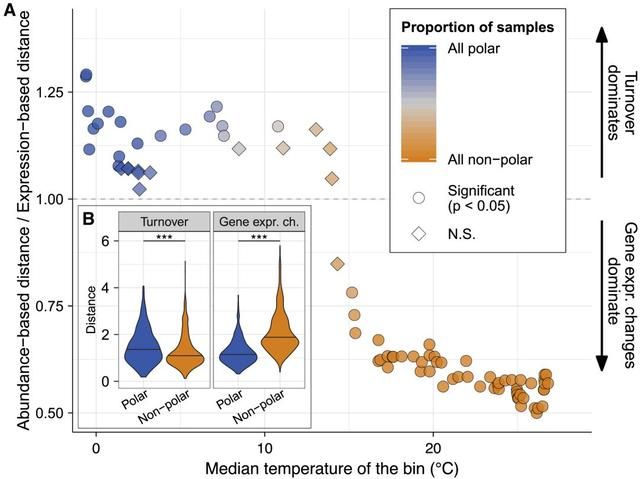
图7 群落更替和基因表达变化对宏转录组组成分变化的相对贡献。(A)通过使用沿温度梯度的滑动窗口将外缘海区样本分成15个样本组,分析该比率与温度的关系。(B)内图表示极地和非极地群落更替和基因表达变化的差异。
最后,作者通过将这些结果从空间离散的数据外推到气候变化的潜在后果,提出假设:微生物群落组成的变化对转录组的相对影响在极地水域将大于非极地水域。然而,这种外推需要在这里分析的数据的限制下加以解释,即它不能解释微生物群落对随时间逐渐变化的进化性适应。因此,需要进一步研究宏转录组在长期时间动态下的变化,来更好地理解环境变化背景下群落更替和基因表达变化的贡献。尽管如此,作者对群落转录组变化的机制进行了首次全球范围的评估,并为今后的工作提供了框架。
结论
大规模的海洋取样考察,例如世界海洋环流实验(WOCE)或GEOTRACES,对我们了解海洋环流、主要营养物质和包括微量金属在内的元素的分布以及它们对气候系统的贡献都是极有价值的。然而,如果没有整合在行星尺度上调节生物地球化学循环的过程,我们对海洋的地球化学和物理知识仍然是不完整的。通过分析环境样本中的全部基因和转录本,我们可以了解在全球范围内驱动这些循环的微生物群落的潜力和活力,从而帮助我们了解通过生物活动形成的海洋物理化学状态的相互交织的过程。
在这项研究中,作者描述了微生物群落转录组组成的全球生物地理模式,并研究了这些组成的变化是如何归因于群落更替和/或基因表达变化的潜在机制。评估构成差异的机制,可以帮助确定微生物群落中活性分子的改变是由基因表达的变化调节的,还是由进化过程中产生的包含基因组修饰的群落更替调节的。此外,对推动群落组成和多样性变化的生态因素的进一步研究,可以帮助更好地预测海洋微生物群落对环境变化的反应。例如,研究者一致认为温度是全球范围内群落水平上基因组、转录组以及物种多样性差异的主要解释因素,尤其在北冰洋地区有广泛的影响(基于目前该地区不成比例的高升温率的预测)。
值得注意的是,这项研究的分析是由一个系统的、因地制宜的、泛海洋的宏基因组和宏转录组数据集所支持的,该数据集与OM-RGC v2一起,补充了现有的为真核生物、原核生物和病毒开发的其他大型数据集。总之,这些将为在生态系统层面上理解海洋浮游生物的多样性、功能和跨生物大小范围的活动铺平道路。为了达到这一目标,整合时间尺度的宏组学数据将非常重要,最好是来自全球的观测数据,以考虑季节变化和其他伴随的环境变化,如海洋的分层、酸化、营养有效性和脱氧。需要这样的协调努力来进一步完善从基因到生态系统的模型,并为环境和气候政策提供信息,这些政策不仅必须考虑微生物如何受到影响,而且还必须考虑微生物如何影响人为气候变化。
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人