Cell | 超深度宏基因组!复原消失的肠道微生物

期刊:Cell
IF:64.5 (Q1)
发表时间:2023.6
研究背景
不同的生活方式会影响微生物组组成,但目前微生物组的研究严重偏向于西方工业化人群,其中工业化人群的特点是微生物群多样性较低。为了理解工业化生活方式引起的微生物组变化,需要结合高深度的宏基因组测序来研究原始人(Hadza)微生物功能、生长动力学和扩散模式。文章利用351份Hadza粪便样本,测序得到9.39T的宏基因组数据,共获得83,044个MAG以及相关生物信息的结果。
实验设计
Hadza是非洲仅存的延续人类祖先觅食传统的种群之一,研究对167个个体进行粪便取样得到351个样本,处理后共388个DNA样本进行测序,同时得到经过纯化的52株细菌分离株(图1A)。根据本研究中新恢复的基因组评估所有样本的微生物组组成,将宏基因组数据映射到一系列定制的数据库中,其中包含细菌/古菌(n= 5,755)、噬菌体(n=68,461)和真核生物(n=12)基因组的物种水平代表的全基因组序列(图2E)。
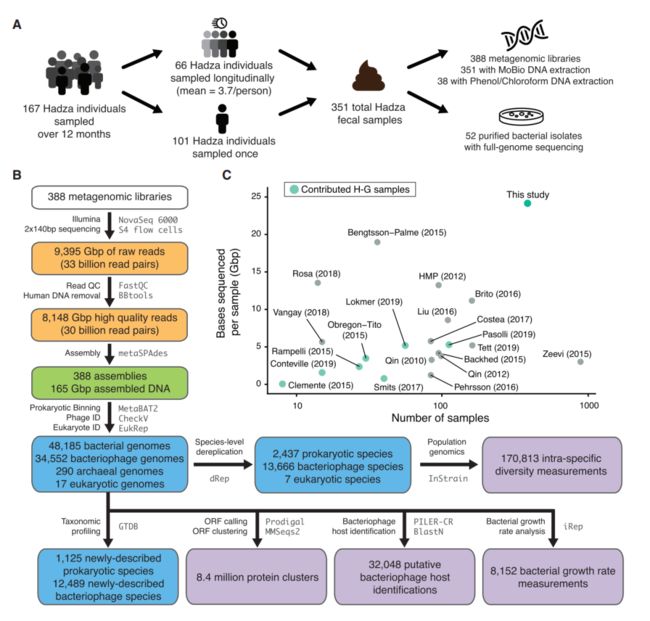
图1 Hadza肠道微生物组数据分析过程
主要结论
1.
Hadza肠道微生物多样性
除了来自UHGG和MGV的数据之外,还可以从Hadza中得到未收录的微生物占总发现微生物比分别为20.2%和79.9%(图2A和2B)。此外,在Hadza肠道微生物中发现了未知的真核生物,共注释到840万个蛋白质家族,其中59.7%不存在于统一人类胃肠蛋白(UHGP)数据库中(图2D)。
将Hadza微生物组与其他生活方式的人群进行比较,对尼泊尔人和加州人的粪便样本进行了深度宏基因组学测序。用这些MAG数据自定义了3个数据库,用基因组数据库来评估微生物组信息(图2E),包括细菌/古细菌(n=5,755),噬菌体(n=68,461), 真核生物(n=12)。Hadza的细菌、噬菌体和古菌的多样性均高于本研究中的其他种群(图2F)。总之,与其他种群相比,不同测序深度的Hadza样本中有更多的细菌、古菌和噬菌体和未知的物种(图2G)。
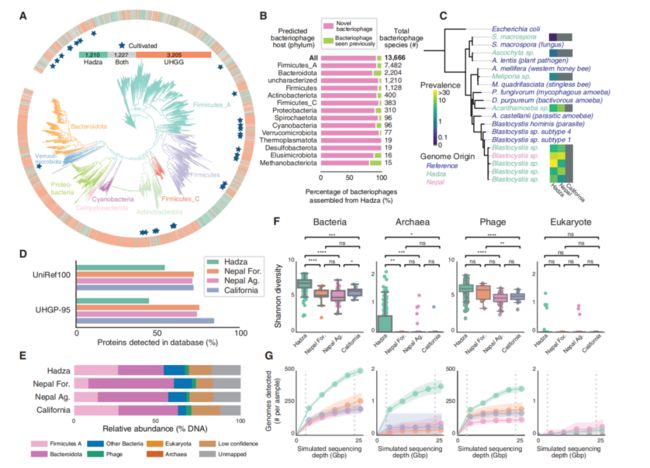
图2 Hadza肠道微生物群包含大量未知物种信息
研究利用稀释曲线来评估随测序深度增加物种的丰富度变化(图 3A)。分析表明,Hadza的平均肠道微生物有730种,而加州人的平均肠道微生物有277种。高深度的测序可以检测低丰度的微生物,所以不同丰度水平下检测到物种的微生物组成不同和具有基因组新颖性(图3B)。
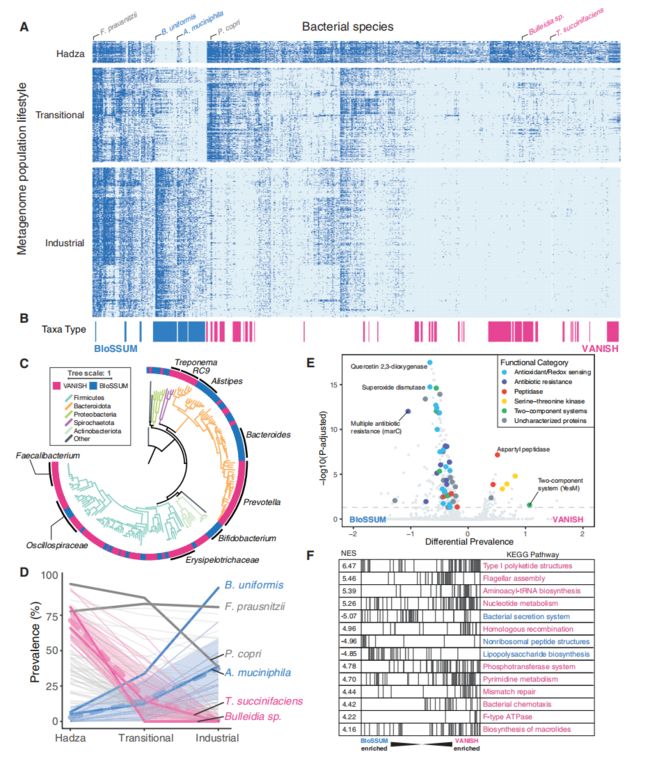
图3 增加测序深度导致检测到未知物种及其系统发育树
2.
Hadza样本中发现在工业化进程中已经消失的肠道微生物种群
本研究通过18篇已发表文献,利用一共1800个数据,包括工业化人群样本950个,过渡人群样本583个,Hadza样本135个和其他狩猎采集者样本132个,根据细菌/古菌在Hadza和工业化种群的富集情况将其分为VANISH(工业化中消失的,124个)和BloSSUM(工业化中受到选择的,63个)两个分类群,进一步分析发现系统发育与工业化进程存在强相关性(图4a-c),在过渡的人类肠道环境中,BloSSUM类群可能比VANISH类群具有竞争优势。
基于个体KEGG分析确定了37和 268个在VANISH和BloSSUM中常见的基因(图4 e)。与VANISH分类群最相关的是肽酶和产孢基因,而BloSSUM类群与抗氧化和氧化还原有关。研究将这些KO聚集到通路中绘制集富集分析图(图4F)。富集的KOs的相关功能和通路的差异表明,与VANISH类群相比,BloSSUM类群在功能上并不冗余。

图4 VANISH 和 BloSSUM 类群具有不同的系统发育和功能
3.
Treponema succinifaciens的分布反映人类的迁移
在Hadza、尼泊尔人和加州人样本上进行的深度测序得到 1,047 个新的 Spirochaetota MAG,MAG的相对丰度随着工业化程度的提高而降低。Treponema Succinifaciens在工业化群体中几乎完全不存在,本研究中发现的 该物种MAGs 数量从 125 个增加到 346 个,并对该物种进行系统进化分析(图 5C),从进化数据观察到Treponema succinifaciens的传播模式与人类迁徙路线一致。

图5 Hadza中高度丰富的螺旋体
4.
Hadza肠道微生物的进化、复制和扩散
采取更深度测序和更新颖的宏基因组方法再次确认了Hadza肠道微生物的组成和碳水化合物活性酶确实存在季节性循环,深度测序首次测量原位复制率在Hadza肠道中也表现出季节性循环。为了阐明Hadza微生物群中具有不同选择压力的基因,对全基因 pN/pS 比率进行了分析,发现存在正向或多样化选择(图 6A)。这可能是由于肠道中的动态变化,如季节性饮食变化、宿主免疫系统和躲避病毒入侵。研究还进行了ANI分析,发现家族个体共享的同源性菌株多于非同源性菌株,比无血缘关系的个体的个体共享更多高度相关的菌株(图 6C)。
图6 Hadza肠道细菌之间的微多样性、复制速率和菌株共享模式
结论
综上所述,通过对Hadza人群粪便进行超深度宏基因组测序,研究结果表明:
1、组装了数千种未知的人类肠道细菌、古菌、真核生物和噬菌体;
2、与生活方式相关的VANISH和BloSSUM分类群的不同功能鉴定;
3、Hadza肠道微生物的复制率变化呈季节性循环,菌株共享模式与其独特的社会结构有关。
参考文献
Carter et al., Ultra-deep sequencing of Hadza hunter-gatherers recovers vanishing gut microbes, Cell 2023,