Rosetta Tutorial 13 ~ 14 翻译 | 主要为 Output Analyzing 及 FoldTree 部分
该文档翻译自Rosetta官网的教程性文档 Rosetta Tutorials, Demos, and Protocol Captures,原文链接:https://www.rosettacommons.org/demos/latest/Home 。“”内的内容为译者添加。
Contents
- 13. Analyzing Rosetta Output
-
- PDB file
- The score file
- Extracting silent files
- Other Points
-
- Clustering
- Controls
- Compare with experimental results
- 14. The Fold Tree: Propagating Changes in the Structure
-
- Internal Coordinates
- The Lever Arm Effect
- Using the Fold Tree
-
- Single Protein
- Complexes With More Than One Chain and Jumps
- Final Points
- 15. Symmetry: Modeling Symmetric Proteins
13. Analyzing Rosetta Output
本教程将通过一些示例介绍如何处理Rosetta产生的输出。通常输出是以下文件中的一个或几个(重要:Rosetta 必然给你一个结果!):
- pdb文件-蛋白质数据库格式的蛋白质结构信息
- 静默文件-蛋白质结构信息在Rosetta自己的格式
- 分数文件-以制表符分隔的分数集
- 日志文件-由给定的运行到终端的所有输出的记录
你要回答的主要问题是:“结果能回答我的问题吗?”
本教程主要关注统计和分析方法,以提供这个问题的答案。与任何随机模拟一样,Rosetta 对任何给定运行产生的许多结果将探索与手头问题无关的构象空间区域;统计分析可以帮助从给定的运行中选择最佳/最有信息的结果,同时还可以从整体上评估运行的成功程度。
PDB file
Rosetta生成的模型通常采用PDB文件格式。如果您打开该文件,您当然会找到所有的原子位置。此外,Rosetta将在pdb文件的末尾附加每个残基的所有评分项。对提供的pdb文件进行评分,并在您最喜欢的文本编辑器中打开评分结构。
$ROSETTA3/main/source/bin/score_jd2.default.linuxgccrelease -s 1ubq.pdb -out:output -out:file:scorefile score.sc
现在,搜索“pose”这个词。这将把你带到坐标部分的最后。它应该看起来像这样:
# All scores below are weighted scores, not raw scores.
#BEGIN_POSE_ENERGIES_TABLE S_0004_design_0062.pdb
label fa_atr fa_rep fa_sol fa_intra_rep fa_elec pro_close hbond_sr_bb hbond_lr_bb hbond_bb_sc hbond_sc dslf_fa13 rama omega fa_dun p_aa_pp ref total
weights 0.8 0.44 0.75 0.004 0.7 1 1.17 1.17 1.17 1.1 1 0.2 0.5 0.56 0.32 1 NA
pose -513.648 51.38 327.036 1.19669 -61.0293 2.86533 -26.313 -42.5744 -9.85907 -17.8907 -11.8656 -9.94805 10.566 150.226 -17.7945 -29.4897 -197.143
THR:NtermProteinFull_1 -2.22882 0.1511 2.60394 0.00934 -0.65245 0 0 0 -0.29914 -0.28075 0 0 0.10172 0.02367 0 0.16454 -0.40685
VAL_2 -2.91409 0.33743 1.30547 0.01945 -0.14243 0 0 0 0 0 0 -0.10417 0.10036 0.61358 0.17219 0.74484 0.13264
| line starts with | What does this mean? |
|---|---|
| label | 能量/打分项的名称 |
| weights | 打分项的权重 |
| pose | 每个所有残基打分项的总和 |
| THR:NtermP[...] | 第一个残基 (在此为N末端苏氨酸)的得分 |
| VAL_2 | 第二个残基 (在此为缬氨酸)的得分 |
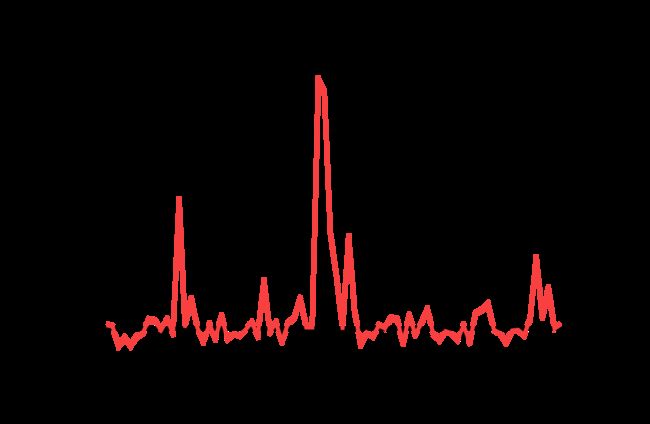
使用egrep提取每个单个残基的总分:
egrep '[^A-Z]*_[0-9]+\s' 1ubq_0001.pdb | cut -d'_' -f2 | awk '{print $1 "\t" $NF}' > total_score_per_res.dat
这将选择包含模式字母_数字的所有行,然后去掉下划线之前的所有内容,最后打印出_后的数字、一个制表符和total_score。现在,您可以绘制剩余位置vs.总分,例如作为直方图。你应该注意到有一些区域能量相对较差(大约在35-40位置)。
The score file
评分文件包含运行中生成的所有模型的评分,分解为各个评分项。您可以提取某些列(即评分项)或按任意列对它们进行排序。下面的命令将按第二列(total_score)进行排序:
sort -n -k2 example_score_file.sc
# 同样按第二列进行排序,但是只输出第2-3行
sort -n -k2 example_score_file | awk '{print $2 "\t" $3}'
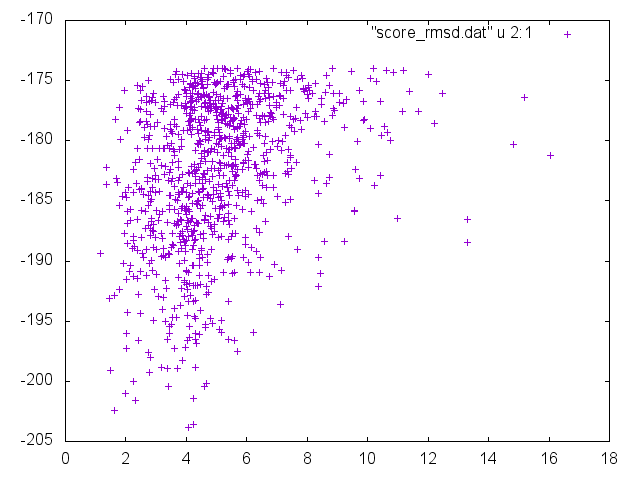
下面这个命令将根据总分排序,只取前1000个,提取列2(total_score)、列25(rmsd)和最后一列(description),并将其写入一个名为score_rmsd.dat的新文件中(注意:得分项在得分文件中的位置取决于您正在运行的 protocol 和您正在使用的选项!):
sort -n -k2 example_score_file.sc | head -n 1000 | awk '{print $2 "\t" $25 "\t" $NF}' > score_rmsd.dat
对score_rmsd.dat的第2列(rmsd)-第1列(total_score)进行绘图。安装gnuplot后,可以很容易地进行快速绘图。但是你可以用任何软件来画出这两列。
gnuplot
Terminal type set to 'x11'
gnuplot> plot "score_rmsd.dat" u 2:1
您可以看到较低的能量值(y轴)对应于较低的rmsds (x-值),这意味着有一个显著的收敛程度。
Extracting silent files
当你产生大量的模型(例如de novo结构预测)时,建议使用下述标签将模型输出为二进制静默文件:
-out:file:silentfile_struct_type binary
-out:file:silent <file name of your choice>
Rosetta提供了多种从这些文件中提取结构的方法。
- 借助评分步骤:评分步骤可以使用沉默文件作为输入,并生成 pdb 文件。
main/source/bin/score_jd2.default.linuxgccrelease -in:file:silent_struct_type binary -in:file:silent example.out -out:pdb -out:file:scorefile extracted_scorefile.sc @general_flags
这将为静默文件中的每个结构提供一个新的打分文件和PDB(这里只有一个结构)。你可以添加-in:file:tags S_00000170_1对某一个结构进行打分提取。
- 使用提取 PDB 的程序:使用
extract_pdbs程序进行提取。首先删除上一步提取的pdb文件然后尝试:
rm S_00000170_1.pdb
main/source/bin/extract_pdbs.linuxgccrelease -in:file:silent_struct_type binary -in:file:silent example.out
-in:file:tags选项在这里也可以使用。你可以通过排序分数文件,找到最优结构,然后使用它们的标签提取以获得最好的能量模型。
Other Points
Clustering
为了得到一个合理可靠的结果,你应该总是得到多个具有相似能量的相似解。但确实会出现人为的高分结构这种异常值。为了找出你的结果是离群值还是在结构和能量上有轻微差异的多次采样,通过结构相似性对模型进行聚类是非常有用的。虽然 Rosetta 有一个聚类应用程序,但它的使用非常有限。其他更复杂的聚类软件应该优先使用。关于聚类的更多信息也可以在 Rosetta 手册中找到。
Controls
评估你是否可以信任 Rosetta 的结果的一个好方法是包括控制运行。通常,你可能会发现同源蛋白、蛋白复合物、肽等与你未知的系统相似,但感兴趣的实验结果是已知的。使用这些作为正向/负向控制是一个好主意。例如,如果已知肽段 TRRTFGAH 可以结合(蛋白或其他),但 Rosetta 无法正确对接,那么您可能不希望信任 TRRSYGAH 的对接结果。相反,如果已知肽 TLLTFGAH 不结合,但 Rosetta 无法区分 TLLTFGAH 和 TRRTFGAH 的(与蛋白或其他的)结合能力,则 protocol 可能不具有对未知亲和肽的结合进行排序的准确性或动态范围。
Compare with experimental results
如果您试图用 Rosetta protocol 预测定量结果,您应该首先基于具有已知结果的类似系统绘制校准曲线。Rosetta 的能量结果是任意单位的,结果对特定 protocol 的精细细节敏感。可能存在相关性(例如(不)稳定性 与 Rosetta 的预测 ddG 之间存在相关性),但相关性的细节是对精确的 protocol 细节敏感的。在具有已知实验结果的系统上运行计算,并查看预测值和实验值之间的相关性将允许您:1. 根据预测结果估算实验值;2. 提供 protocol 预测精度的判断。一个非常嘈杂的相关性意味着预测的准确性是有问题的,此时你应该对 Rosetta 产生的预测持怀疑态度。
14. The Fold Tree: Propagating Changes in the Structure
KEYWORDS: CORE_CONCEPTS GENERAL
Tutorial by Parisa Hosseinzadeh ([email protected]). Edited by Sebastian Rämisch ([email protected]). File created 21 June 2016 as part of the 2016 Documentation eXtreme Rosetta Workshop (XRW).
在本教程中,您将了解折叠树、内部坐标的概念,以及如何明智地使用它们来获得有意义的输出。
Internal Coordinates
如果您打开一个PDB文件并查看它,您可以看到它包含所有原子的信息及其在3D空间中的 (x, y, z) 坐标。当你想移动一个蛋白质时,你可以改变每个原子的3个坐标。换句话说,每个原子有3个自由度。然而,3个自由度对于我们的计算来说太大了。为了使事情更简单,Rosetta 使用内部坐标。也就是利用它与相邻原子之间的键,角度和扭力定义一个原子。当你使用残基的键、角和扭转角值,而不是 x、y、z 值来描述一个原子时,你使用的是它的内部坐标。
在内部坐标世界中,你通过改变键、角度和扭转来移动物体,所以还是有3个自由度。然而,这些自由度并不同等重要。对于大多数蛋白质的建模,键的长度和角度变化很小——我们可以有效地认为它们是固定的。运动主要是通过扭转角度的变化发生的。因此,我们将自由度从每个原子3个减少到每个原子1个。此外,对于某些原子(像大多数氢原子),它们的扭转自由度是由它们的化学环境所决定的。这些原子的自由度从3减少到0。
你可以在ICOOR_INTERNAL开头的行显示了残基中原子的内部坐标(请参阅这里了解ICOOR_INTERNAL行中每一列的含义)。
The Lever Arm Effect
当你改变一组给定余数的扭转角度但你把其余的设为不动,会发生什么?让我们看看下面这个图:
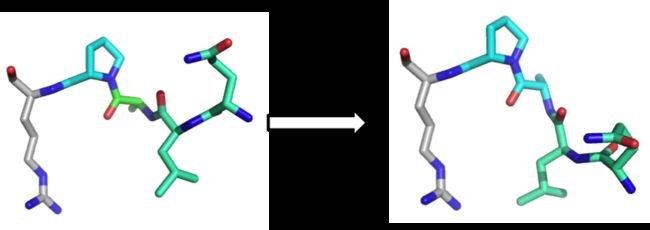
你可以看到为了改变一个特定的扭转角度,在蓝色残基中所有在这个改变的原子右边的都应该移动。换句话说,尽管它们保持相对于对方固定,它们仍然一起移动以适应下游的蓝色残基的变化。
现在让我们看看同样的例子,但有一点不同:
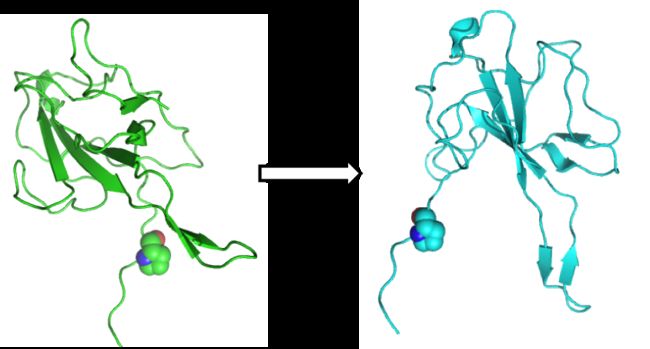
你可以看到蛋白质n端一个扭转角度的小变化会引起蛋白质其余部分的巨大移动。这被称为杠杆臂效应。我们如何避免这种情况呢?
Using the Fold Tree
Single Protein
在这一部分中,我们将看到如何使用折叠树来控制蛋白质中各部分的运动。折叠树是一种告诉 Rosetta 给定结构中残基之间连通性的方法。它定义了哪些残基是上游或父残基,哪些残基是下游或子残基。让我们通过一些例子来把事情弄清楚。在这个例子中,你将使用冠状病毒核衣壳N端部分的结构。在教程中使用这个命令导航到折叠树目录:
cd <path_to_Rosetta_directory>/demos/tutorials/fold_tree
打开输入目录中为您提供的capsid.pdb,你可以看到它有一个长而无结构的N末端,其余的结构大部分是β片层。我们知道N端区域是无序的,所以在处理PDB之前,我们想要弛豫这部分。cps_relax.xml是提供给您的一个 Rosetta 脚本,它使用movemap弛豫结构的残基1-20。
...
<MoveMap name="part">
<Span begin=1 end=20 bb=1 chi=1/>
</MoveMap>
...
这里用到了 Rosetta 默认的折叠树caps_tree1.ft:
FOLD_TREE EDGE 1 133 -1
其含义为:从1到133的EDGE是聚合EDGE(-1)。从A链的残基1开始,一直到最后一个残基(在这个例子中是133),任何上游残基(接近N端的残基)的移动都会导致下游残基(更接近C端)的移动。这条链是通过共价键以线性方式连接的不间断连续残基簇。我们称这个集群为EDGE。-1表示EDGE中的残基通过共价键连接。在fold_tree/文件夹中,使用下面的命令运行脚本,弛豫残基1-20,而不改变结构其余部分的主干:
<path_to_Rosetta_directory>/main/source/bin/rosetta_scripts.default.linuxgccrelease -in:file:s inputs/capsid.pdb -parser:protocol inputs/caps_relax1.xml -out:prefix test1_
在1-10分钟之后,您可以看到生成了输出test1_capsid_0001.pdb。现在将原始结构与输出进行比较,你可以看到N端部分与原来的结构相比有了很大的变化。如果我们只对齐N端部分,你可以看到蛋白质的其余部分已经剧烈地移动,以适应N端前20残基的变化。
现在,让我们看看当我们交换下游和上游的位置,告诉 Rosetta 运动应该从C端传播到N端,会发生什么。这在caps_tree2.ft中将折叠树倒置:
FOLD_TREE EDGE 133 1 -1
现在再运行一轮:
<path_to_Rosetta_directory>/main/source/bin/rosetta_scripts.default.linuxgccrelease -in:file:s inputs/capsid.pdb -parser:protocol inputs/caps_relax2.xml -out:prefix test2_
这应该不会超过10分钟。看看test2_capsid_0001.pdb输出结构并与capsid.pdb和test1_capsid_0001.pdb的结构比较,你现在可以看到蛋白质的移动要小得多。这是因为现在C端部分在变化发生的地方的上游,并且不会因为N端移动而移动。因此,如果你有一个具有较大摆动性的N末端的蛋白质,它会在你的运行过程中移动很多,你可能需要因此改变折叠树。
Complexes With More Than One Chain and Jumps
现在让我们运行另一个例子。在这个例子中,我们运行一个假设的泛素二聚体ubq_dimer.pdb。让我们看看如果弛豫这个姿态会发生什么。在ubq_relax1.xml Rosetta 脚本中,我们使用movemap来固定除了C终端中小loop外的主干。在第一次运行中,我们将使用 Rosetta 的默认折叠树来处理蛋白质复合物。让我们来看看包含这个折叠树的ubq_tree1.ft:
FOLD_TREE EDGE 1 76 -1 EDGE 76 77 1 EDGE 77 152 -1
它的意思是:从1到76的EDGE是蛋白质的EDGE(-1);从76到77的EDGE是第一个jump EDGE(1);从77到152的EDGE是蛋白质的EDGE(-1)。你可以看到我们有三条边:第一个应该很熟悉,它说第一条边从残基1开始,通过共价键经过残基76(基本上是链A);第三条边是相同的含义,但对链B(注意这些是姿态编号)。现在,我们来看看第二条边。你可以看到它从A链的最后一个残基(76)到B链的第一个残基(77),这不是共价相互作用,但是,因为 Rosetta 使用内部坐标(扭转角),而不仅仅是3d坐标,所以结构中的所有部分都必须连接起来;如果没有共价连接(就像我们这里的例子),链必须与一个我们称为 JUMP 的假想链接连接。跳跃本身就是一条EDGE。在折叠树中,跳跃显示为正数。第一次跳跃得到数字1,每次跳跃数字都会递增。
现在让我们使用默认的折叠树来弛豫:
/main/source/bin/rosetta_scripts.default.linuxgccrelease -s inputs/ubq_dimer.pdb -parser:protocol inputs/ubq_relax1.xml -out:prefix test1_
1-5分钟后,您应该会得到一个名为test1_ubq_dimer_0001.pdb的输出。把它和原来的二聚体比较一下,你可以看到第二条链从原来的位置移动了很多,这是杠杆臂效应的另一个例子。A链C末端最后几个残基的运动会一直传播到整个A链。
现在,让我们看看如何解决这个问题。假设我们根据实验知道A链中的 Val71 和B链中的 Val146 在界面形成中很重要,并希望确保它们在弛豫过程中保持紧密。所以,我们可以重新定义我们的折叠树,让残基71和146是直系父结点,或者说是最上游的。看看修改后的折叠树inputs/ubq_tree2.ft:
FOLD_TREE EDGE 71 1 -1 EDGE 71 76 -1 EDGE 71 146 1 EDGE 146 77 -1 EDGE 146 152 -1
和上面讲的一样,这是一个蛋白质边(-1),蛋白质边(-1),跳跃边(1),蛋白质边(-1),蛋白质边(-1)。你可以看到现在有多个边。A链现在由两条边定义:第一条边从残基71一直到A链的N端,第二条边从残基71到A链的C端;JUMP 现在是两个父残基71到146;链B也是由两条边定义的。下图显示了这两棵折叠树的不同之处:

红色虚线表示跳跃,箭头表示每个边的方向。现在,让我们再次运行,但这次是使用我们刚刚定义的新折叠树:
/main/source/bin/rosetta_scripts.default.linuxgccrelease -s inputs/ubq_dimer.pdb -parser:protocol inputs/ubq_relax2.xml -out:prefix test2_
看看test2_ubq_dimer_0001.pdb输出并与原始的比较。你可以看到A链的C端弛豫导致的B链的剧烈运动现在减弱了。
Final Points
现在你知道什么是折叠树、它是如何用来控制结构中的运动的以及如何使用它。你也可以用折叠树来说明一个结构中不同的链是如何相互移动的。例如,如果你的链C在一个复杂的链A和B之间,它的运动应该影响链B,你可以使用一个折叠树,将链C放在链B的下游。同样的原理适用于任何链,包括配体或金属。所以,你可以通过应用不同的折叠树来控制它们的运动行为。如果你对对称姿势的折叠树设置感兴趣,请点击这里。请注意,折叠树必须不出现循环,换句话说,你不能同时把一个残基定义为另一组残基的上游和下游。