GROMACS Tutorial 5: Protein-Ligand Complex 中文实战教程
GROMACS Tutorial 5: Protein-Ligand Complex 中文实战教程
- 前言
- 系统环境
- 特别强调
- 一、预处理阶段
-
- 1.1 蛋白质配体分离以及除水操作
- 1.2 选择力场识别JZ4配体
-
- 1.2.1 使用在线力场解析
- 1.2.2 使用官方推荐力场CHARMM36解析
- 1.3 蛋白的top文件准备
- 1.4 配体的top文件准备
- 1.5 使用CgenFF生成配体top文件
- 1.6 组合蛋白质和配体,修改生成的gro文件和top文件,生成complex文件
-
- 1.6.1 修改gro文件
- 1.6.2 修改top文件
- 二、添加离子
-
- 2.1 定义盒子并添加溶剂
- 2.2 添加离子准备
- 2.3 添加离子
- 三、能量最小化
- 四、限制配体及体系平衡
-
- 4.1 限制复合物
- 4.2 nvt平衡
-
- 4.2.1 nvt平衡设置
- 4.2.2 nvt画图
- 4.3 npt平衡
-
- 4.3.1 npt平衡设置
- 4.3.2 npt画图
- 五、成品模拟
- 六、分析
-
- 6.1 分析准备工作
- 6.2 能量分析
- 总结
前言
因为我觉得GROMACS官方英文教程和李继存老师的GROMACS的中文教程对于新手非常不友好,并且有很多细节的地方并没有详细说明,导致有很多困难的地方。这里记录一下自己成功的实验案例。
书接上回,我们做完了GROMACS Tutorial 1,想必已经对基本操作有了足够的熟悉,这次我们来做蛋白质与配体的复合物教程。
提示:以下是本篇文章正文内容,下面案例可供参考
系统环境
这个和GROMACS Tutorial 1保持一致
电脑系统:win11(大部分操作) linux:ubuntu22.04 (小部分操作)
我知道gromacs很多在linux上搞,但我就要在windows上搞,windows可以很方便的可视化
python环境:3.6.13
大家还是最好创建一个新的conda环境,我以前就是用的3.11的python结果很多地方报错,最好还是和我用的版本一样
gromacs版本:gmx2020.6_AVX2_CUDA_win64
英文官方教程推荐using a GROMACS version in the 2018.x series,别用老的,老的以前不能使用gpu加速,之后处理会非常慢,除非你用超算,但这就不属于个人电脑能处理的,我是希望能在自己的电脑上跑出来
GPU:GTX1060 6G
相当老的甜品级显卡了,不要用AMD的显卡就行
重要软件:VSCode
我们很多操作都在用VSCode打开并且修改,必须要有,vscode里也装上gromacs的插件,更好看一点
其他软件:pymol3.8 VMD Avogadro(1.4配体准备时需要加氢操作会用到Avogadro)
如果环境不会搭的话我会再出一期专门配置环境的,环境配置并不是我们这次教程的重点
这章GROMACS英文教程地址:[GROMACS英文教程地址](http://www.mdtutorials.com/gmx/lysozyme/index.html)
特别强调
我们所有在的命令操作都要在工作目录中进行!
一、预处理阶段
我先放英文教程,再说我的。

1.1 蛋白质配体分离以及除水操作
下载T4溶菌酶 3HTB 这个是蛋白质和配体一起,我们第一步还是除水。
![]()
用pymol打开3HTB,可以看到最中间的就是配体部分,在GROMACS Tutorial 1我们用的VsCode进行除水,今天再介绍另一种方法,用pymol进行除水。
用pymol打开3HTB,外围有很多红色的小分子,都是水

点击3htb的Action(A)选项选择remove waters,再点击右下角S,显示序列信息。

处理完之后发现上方显示序列信息并且水已经除去。
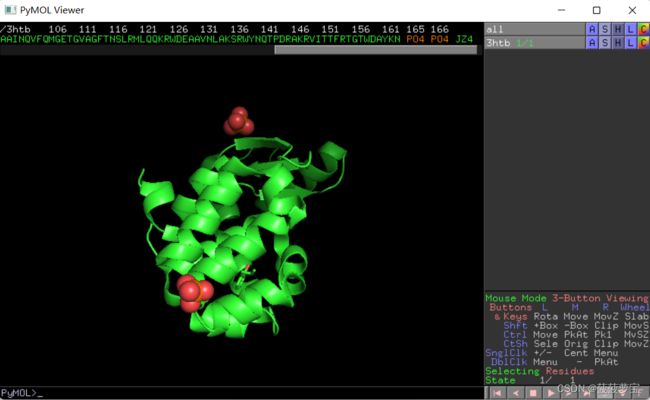
并且发现166之后有个JZ4的配体文件。我们点击JZ4,选择将其摘出。
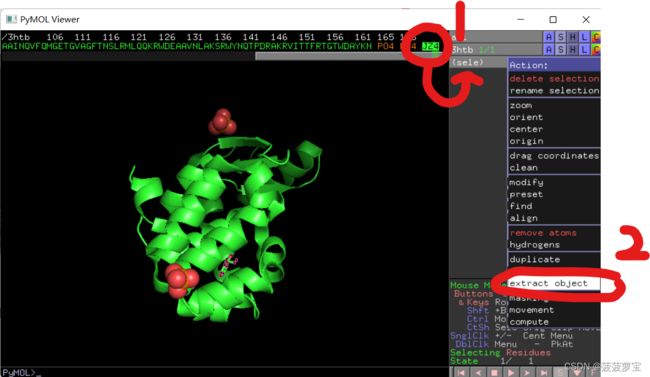
这下生成obj01,这个我们摘出的JZ4配体

之后我们针对原先3THB蛋白,删除除了蛋白质以外的部分。就是删除P04和JZ4,只保留蛋白。因为JZ4被摘除了,所以刚刚JZ4的地方显示为BME
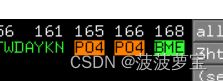
点击P04,P04,BME之后,鼠标右键选择remove。
这样我们就得到了一个蛋白质文件和一个配体文件

然后将两个文件分别保存。
依次保存蛋白质文件和配体文件到工作目录!并且分别命名为3thb_myclean.pdb和jz4.pdb文件(注意文件命名,之后操作要与文件名对应)

得到两个文件

1.2 选择力场识别JZ4配体
因为GROMACS提供的所有力场都不能识别JZ4配体,所以需要采用推荐力场
两种方式 1、采用在线 2、采用官方力场
1.2.1 使用在线力场解析
针对1.2.1使用在线力场解析,我这里不多讲解,有兴趣的同学可以自己研究一下。我着重会讲解官方推荐的方法,在1.2.2里。
由于力场无法自动处理配体,我们需要第三方的方法获得配体的力场参数,使用ACPYPE在线版处理配体
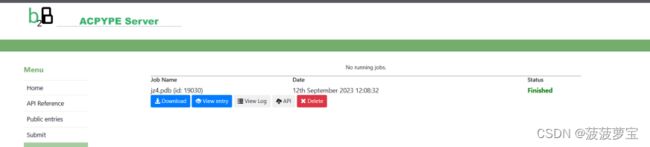
上传jz4.pdb文件得到

1.2.2 使用官方推荐力场CHARMM36解析

点击MacKerell lab website,选择for GROMACS

下载力场及相应的py文件

我选择的是charmm36_ljpme-jul2022.ff.tgz力场文件,因为我的python版本是3.8,所以我选择的是cgenff_charmm2gmx_py3_nx2.py脚本文件。
将下载好的力场文件和python文件放进工作目录中,并且将力场文件进行解压。
这是下载好的python文件
![]()
力场压缩版解压后得到一个文件夹。
![]()
这里的文件夹和python文件都要放在工作目录中
1.3 蛋白的top文件准备

进入控制台,进入工作目录。(工作目录cmd或者conda promp进工作目录)
gmx pdb2gmx -f 3HTB_clean.pdb -o 3HTB_processed.gro -ter
这里的文件要与1.1保存的文件一致,比如我刚刚保存的蛋白质文件叫3HTB_myclean.pdb,那么执行代码也要相应的变化。
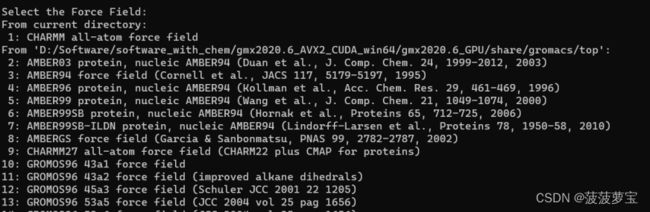
选择刚刚装上的力场 1
选择1回车
之后选择水模型 选择TIP3P
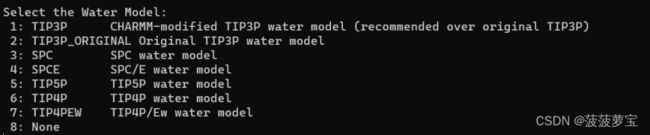
选择1回车
之后选择终端类型 选择NH3+
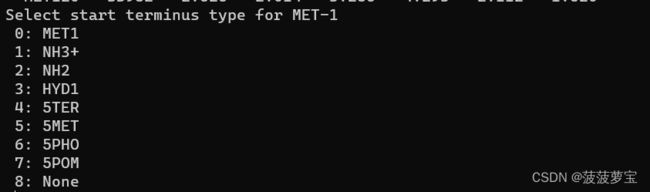
选择1回车
之后选择羧基端类型 选择COO-

选择0 选择COO-
生成结构文件gro 拓扑文件top 位置限制文件itp
 蛋白质准备结束
蛋白质准备结束
1.4 配体的top文件准备
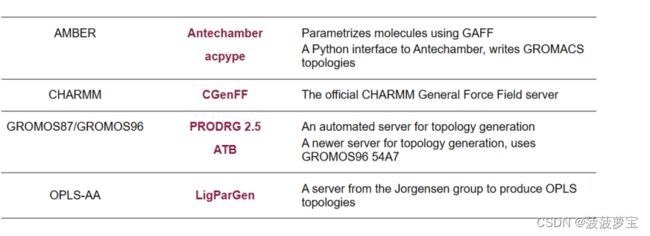
我们的力场选择了官方推荐的charmm36力场,自然选择后续的CGenFF(The official CHARMM General Force Field server)
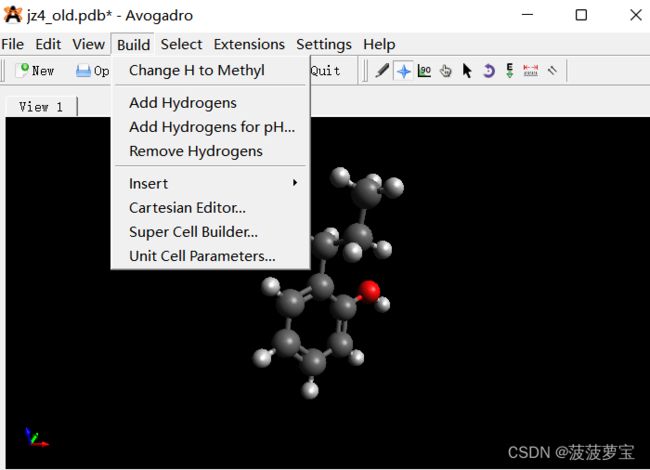
但是使用CgenFF之前要加氢 使用Avogadro来进行加氢
使用Avogadro打开我们的jz4.pbd文件,选择Add Hydrogens
选择完成后File->Save as保存成mol2格式。
保存mol2格式,加完氢的文件得到
![]()
用vscode打开

这个文件是不行的,配体名字没有,只有原子信息。我们需要修改他。
在vscode中在工作目录创建一个新的文件,命名为jz4_h_jz4.mol2,将刚刚的jz4_h.mol2的文件内容全部复制粘贴过去。
修改我画圈的地方(1、将*******改成配体名字JZ4;2、将LIG1改成配体名字JZ4)
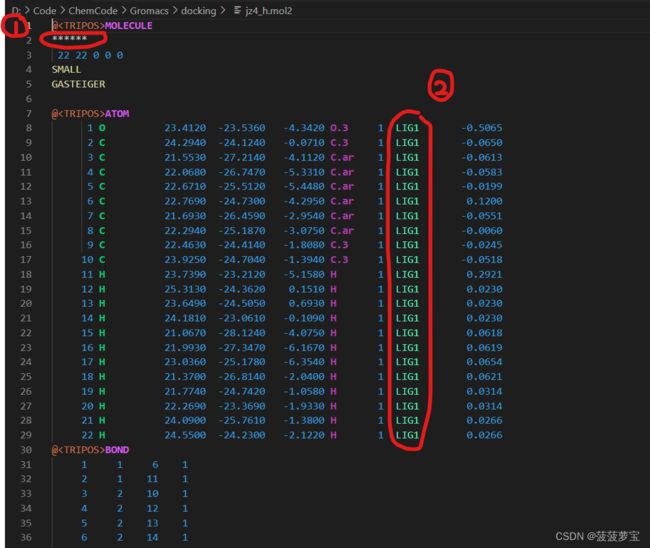
修改好的jz4_h_jz4.mol2文件如下

保存jz4_h_jz4.mol2文件
![]()
对于修改好的jz4_h_jz4.mol2要进行键的修饰
使用sort_mol2_bonds.pl进行修饰,点击这里是文件信息,将其复制粘贴之后,进入linux中,上传我们修改好名字的jz4_h_jz4.mol2文件和sort_mol2_bonds.pl,在linux终端中运行
perl sort_mol2_bonds.pl jz4_h_jz4.mol2 jz4_fix.mol2
![]()
得到文件jz4_fix.mol2文件,将其从linux文件中传回windows里的工作目录。
![]()
将这个文件上传到CgenFF进行,点击这里是CgenFF的网址
登录后选择最上方uploda molecule
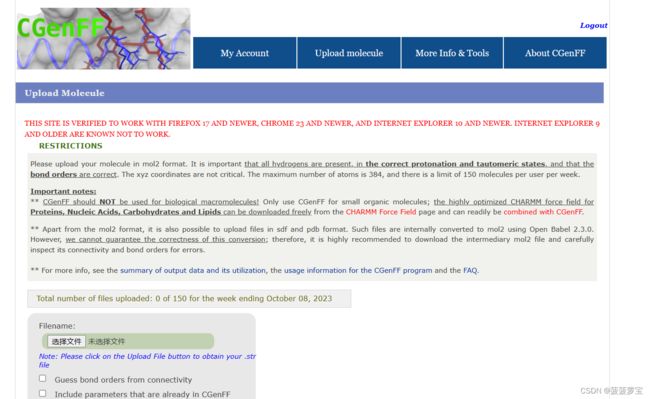
上传我们已经修饰好键的mol2文件。

cgenff从mol2文件生成str文件,将其下载,用vscode打开

生成的str文件是CgenFF自己给你的top文件进行打分,如果小于10则cgenff认为你这个top很不错 10-50凑活 大于50不能用
可以用cgenff来检查自己的蛋白质复合物的配体的情况
1.5 使用CgenFF生成配体top文件
py脚本使用![]()
配体立场使用![]()
python cgenff_charmm2gmx.py JZ4 jz4_fix.mol2 jz4.str charmm36-jul2022.ff
这里的JZ4一定是在1.4里你自己修改的*******那里的名字。一定要对应上,否则gromacs就会报错。
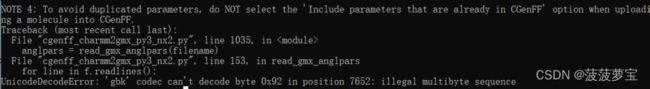
第一次运行报错,提示gbk不能被识别,这是我们python脚本有问题,修改一下编码格式。
打开我们的cgenff_charmm2gmx_py3_nx2.py脚本文件。
将里面所有的f = open(str_filename, 'r')改为f = open(str_filename, 'r',encoding='utf-8')
注意是所有的f = open(str_filename, 'r')改为f = open(str_filename, 'r',encoding='utf-8')!!!!
修改好后再进行执行,成功

生成的文件如下:拓扑文件top 位置限制文件itp

gmx editconf -f jz4_ini.pdb -o jz4.gro
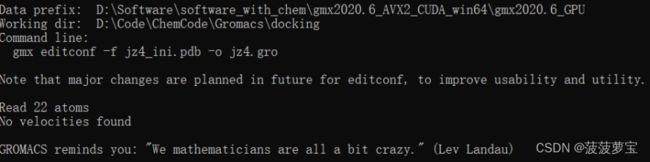
生成结构gro文件和拓扑文件top

1.6 组合蛋白质和配体,修改生成的gro文件和top文件,生成complex文件
1.6.1 修改gro文件
针对上一步生成的3HTB_processed.gro(1.3生成的)和这一步生成的jz4.gro
要将jz4粘贴进去生成新的complex.grp(先复制一份3HTB_processed.gro,再把jz4.gro加进去)
1.3生成的3HTB_processed.gro用vscode打开如下,注意第二行2614显示原子个数
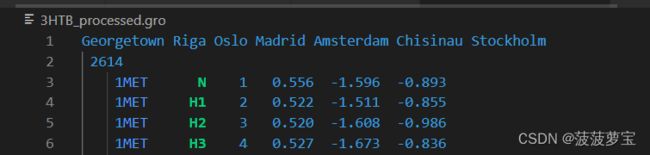
1.5生成的jz4.gro用vscode打开如下,注意第二行22显示原子个数
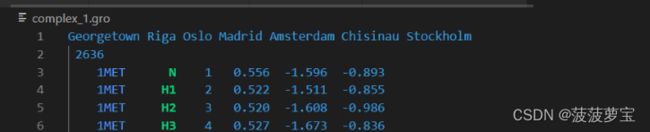
先复制一份3HTB_processed.gro命名为complex_1.gro,再把jz4.gro加进去,加到最后一行之前即可,修改好的complex_1.gro文件如下
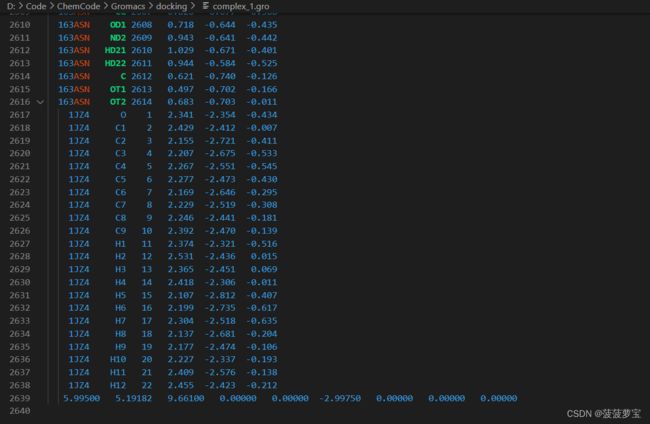
针对已经添加的jz4.gro的complex_1.gro文件,还要修改在第二行修改原子个数,complex_1.gro的原子个数等于3HTB_processed.gro的原子个数+jz4.gro的原子个数,也就是2614+22=2636
把complex_1.gro的原子个数改为2636
1.6.2 修改top文件
针对1.5生成的topol.top和jz4.itp文件
在前面环境说明条件中增加条件jz4.ptm文件

在生成的topol.top文件的水模型前加配体文件

在最后一行添加配体文件

保存top文件,准备工作结束。
二、添加离子
2.1 定义盒子并添加溶剂
定义12面体的盒子
gmx editconf -f complex_1.gro -o newbox.gro -bt dodecahedron -d 1.0
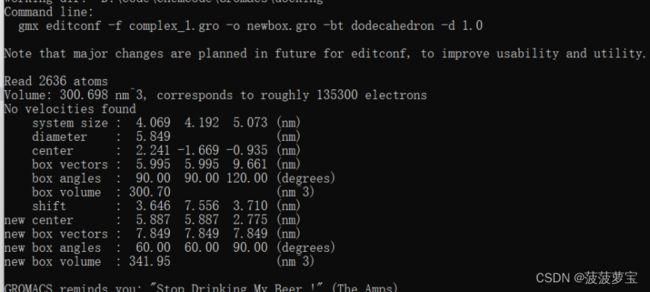
生成
![]()
往盒子中添加溶剂
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro

前后top文件对比


多出了SOL溶剂
生成

理论上应该是十二面体并且蛋白质在最中间
可视化有些问题,不过无伤大雅

2.2 添加离子准备
第一步生成蛋白质top时复合物带6个正电荷
先生成配置mdp文件,在工作目录中用vscode创建ions.mdp文件,点击这里复制粘贴进ions.mdp并保存。
![]()
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr

生成

2.3 添加离子
官方给的代码,这个不可以复制粘贴直接使用,必须根据相应力场文件里的ions.itp中找相应离子缩写,这个我在GROMACS Tutorial 1的3.3说的很详细了,可以去回看一下。GROMACS Tutorial 1
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral
在charmm36力场里,钠离子叫SOD,氯离子叫CLA,因此我们的代码应该改成
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname SOD -nname CLA -neutral

选择15 SOL进行包埋离子,离子要替换掉的对象一定是溶剂
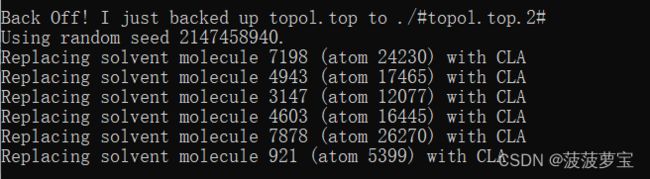
生成

查看top文件,topol文件中已经加入cla离子
因为蛋白质带6个正电荷,所以要补6个氯离子

用pymol可视化一下
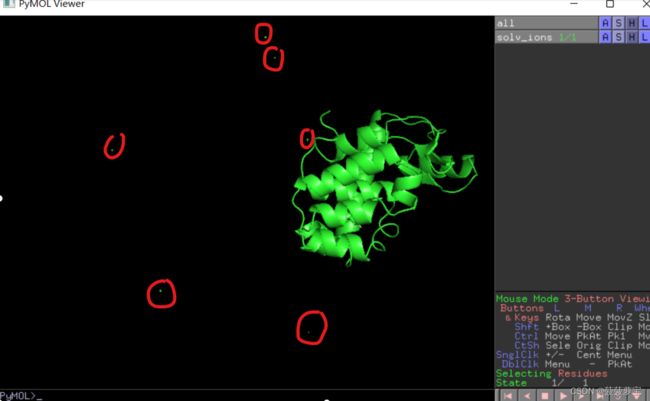
肉眼可见添加了六个cl离子
三、能量最小化
准备配置文件em.mdp,先生成配置mdp文件,在工作目录中用vscode创建em.mdp文件,点击这里复制粘贴进em.mdp并保存。
![]()
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr
生成

gmx mdrun -v -deffnm em

生成

四、限制配体及体系平衡
4.1 限制复合物

gmx make_ndx -f jz4.gro -o index_jz4.ndx

输入
0 & ! a H*
再输入
q
生成
![]()
gmx genrestr -f jz4.gro -n index_jz4.ndx -o posre_jz4.itp -fc 1000 1000 1000
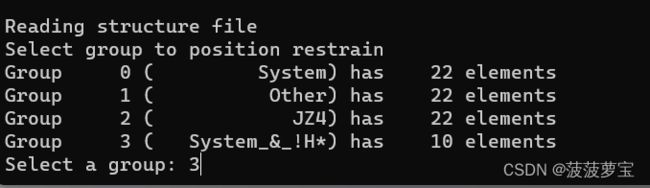
选择3
修改top文件
gmx make_ndx -f em.gro -o index.ndx

先组合蛋白和配体
选择1|13
还需要把水和离子组合一下
选择14|15
再选择q,这里生成的组合很重要!!!后面要用!!!!!
蛋白质配体组合叫Protein_JZ4
水离子组合叫CLA_Water

生成
![]()
4.2 nvt平衡
4.2.1 nvt平衡设置
准备配置文件nvt.mdp,先生成配置mdp文件,在工作目录中用vscode创建nvt.mdp文件,点击这里复制粘贴进nvt.mdp并保存。
这里不能直接用官方文档给的代码,要将里面的tc-grps修改成我们刚刚组合的东西。蛋白质配体组合叫Protein_JZ4,水离子组合叫CLA_Water
![]()
改成
![]()
之后保存
![]()
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tpr
生成

gmx mdrun -v -deffnm nvt

生成

4.2.2 nvt画图
gmx energy -f nvt.edr -o temperature.xvg

选16 0
![]()
python画图(具体代码在GROMACS Tutorial 1里详细讲过,可以回看一下)

4.3 npt平衡
4.3.1 npt平衡设置
准备配置文件npt.mdp,先生成配置mdp文件,在工作目录中用vscode创建npt.mdp文件,点击这里复制粘贴进npt.mdp并保存。
这里不能直接用官方文档给的代码,要将里面的tc-grps修改成我们刚刚组合的东西。蛋白质配体组合叫Protein_JZ4,水离子组合叫CLA_Water
改成

gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr
生成

gmx mdrun -v -deffnm npt
生成

4.3.2 npt画图
gmx energy -f npt.edr -o pressure.xvg
 选17 0
选17 0
![]()
python画图

五、成品模拟
准备配置文件md.mdp,先生成配置mdp文件,在工作目录中用vscode创建md.mdp文件,点击这里复制粘贴进npt.mdp并保存。
这里不能直接用官方文档给的代码,要将里面的tc-grps修改成我们刚刚组合的东西。蛋白质配体组合叫Protein_JZ4,水离子组合叫CLA_Water
改成

生成
![]()
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_10.tpr
生成

gmx mdrun -v -deffnm md_0_1 -nb gpu


gtx1060预计从18点45跑到22点01,预计跑4个小时
生成

六、分析
6.1 分析准备工作
消除周期性
gmx trjconv -s md_0_10.tpr -f md_0_10.xtc -o md_0_10_center.xtc -center -pbc mol -ur compact

选择1作为体系进行偏移
 选择0为输出结果
选择0为输出结果
生成
![]()
因为蛋白质复合物涉及许多跨越周期边界,因此需要确定一个定心的自定义索引组
gmx trjconv -s md_0_10.tpr -f md_0_10_center.xtc -o start.pdb -dump 0
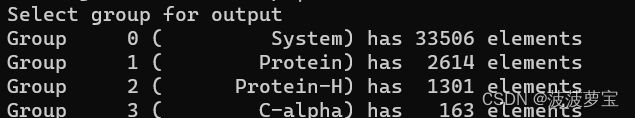
选择0
生成
![]()
为了更平滑的可视化,执行旋转和平移拟合
gmx trjconv -s md_0_10.tpr -f md_0_10_center.xtc -o md_0_10_fit.xtc -fit rot+trans
生成
![]()
6.2 能量分析
准备配置文件ie.mdp,先生成配置mdp文件,在工作目录中用vscode创建ie.mdp文件,点击这里复制粘贴进ie.mdp并保存。
生成
![]()
gmx grompp -f ie.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o ie.tpr
生成
gmx mdrun -v -deffnm ie -rerun md_0_10.xtc -nb cpu
生成

gmx energy -f ie.edr -o interaction_energy.xvg
这个只是示例,根据你选的选项生成不同图,比如the short-range Lennard-Jones energy(LJ-SR)或者The average short-range Coulombic interaction energy(Coul-SR)


gmx energy -f ie.edr -o LJ_SR.xvg
选择22 0

能量为-99±7.0 kj/mol对应官方文档the short-range Lennard-Jones energy为-99.1±7.2 kj/mol

gmx energy -f ie.edr -o coul_SR.xvg
选择 21 0
 能量为-19±7.8kj/mol对应官方文档The average short-range Coulombic interaction energy为-20.5±7.4kj/mol
能量为-19±7.8kj/mol对应官方文档The average short-range Coulombic interaction energy为-20.5±7.4kj/mol

总结
这就是GROMACS Tutorial 5: Protein-Ligand Complex 中文实战教程的所有内容啦,因为我的专业是软件工程,gromacs有时候科研的时候会用到,踩了很多坑,希望学弟学妹们就别绕远路了。欢迎大家在我的评论区留言讨论。