Prodigal-原核生物基因预测
文章目录
- Prokaryotic gene feature
- What is Prodigal?
- Installing Prodigal
-
- Installing on Mac OS X
- Installing on Generic Unix
- Installing on Windows
- Usage
- Advice By Input Type
-
- Finished Genomes
- Draft Genomes
- Metagenomes
- Alternate Genetic Codes
- Organisms with Gene Decay
- Plasmids, Phages, Viruses, and Other Short Sequences
- Understanding the Prodigal Output
-
- Gene Coordinates
- Protein Translations
- Nucleotide Sequences
- 早期版本:Gene Prediction Modes
- Normal Mode
- Anonymous Mode
- Training Mode
- 耗时
- 统计全长基因
- 参考
Prokaryotic gene feature
- 原核生物mRNA的5’端无帽子结构,3’端没有或只有较短的多聚(A)结构
- 原核生物起始密码子AUG上游有一被称为Ribosome Binding Site (RBS)或SD序列(Shine –Dalgarno sequence)的保守区,该序列与16S-rRNA 3’端反向互补,被认为在核糖体-mRNA的结合过程中起作用
- 原核生物常以AUG(有时GUG,甚至UUG)作为起始密码子;真核生物几乎永远以AUG作为起始密码子
- 半衰期短:原核生物中,mRNA的转录和翻译是在同一个细胞空间里同步进行的,蛋白质合成往往在mRNA刚开始转录时就被引发了;大多数细菌mRNA在转录开始1分钟后就开始降解
- 许多原核生物mRNA以多顺反子的形式存在:原核细胞的mRNA(包括病毒)有时可以同时编码几个多肽
单顺反子mRNA (monocistronic mRNA):只编码一个蛋白质的mRNA
多顺反子mRNA(polycistronic mRNA):编码多个蛋白质的mRNA
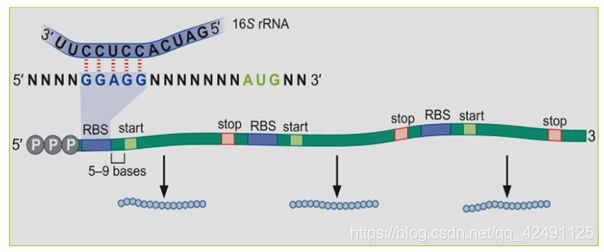
图1. 原核基因结构,显示基因起始位点上游核糖体结合位点(RBS),同一个基因同时编码生成3个肽段(多顺反子)
What is Prodigal?
Prodigal是为细菌和古菌基因组进行蛋白编码基因预测的软件,其缩写源于PROkaryotic DYnamic Programming Genefinding ALgorithm,表示原核生物基因预测的动态规划算法
最早在2007年,在美国能源部联合基因组研究所(DOE)的支持下,由橡树岭国家实验室和田纳西大学诺克斯维尔分校联合开发.2010年,发表在BMC Bioinformatics.很快,Prodigal成为使用最多的微生物基因预测算法之一。截至2021年2月,该出版物已被引用4400多次,在NCBI的ftp网站上保存着使用Prodigal预测的所有细菌和古生菌基因组的结果。
Prodigal可以做的:
- 预测蛋白编码基因并输出多种格式:Prodigal以GFF3、Genbank或Sequin表格的形式提供快速、准确的蛋白编码基因预测;
- 可同时处理draft genomes和metagenomes:在finished genomes, draft genomes, 以及metagenomes上都可以进行基因预测;
- 运行速度快:Prodigal在MacBook Pro电脑上10秒钟就能分析出E. coli K-12基因组的蛋白序列;
- 无监督运行:Prodigal是一种无监督的机器学习算法,它不需要提供任何训练数据,而是从序列本身自动学习基因组的特性,包括遗传密码使用,核糖体结合位点(RBS)的motif;
- 可以处理基因组gaps, scaffolds, and partial genes: 用户可以指定Prodigal应该如何处理gaps,允许或禁止gap区域的基因预测;
- 识别翻译起始位点:Prodigal可以预测大多数基因的正确翻译起始位点,并可以输出基因组中每个潜在起始位点的信息,包括置信度、RBS motif等;
- 输出每个基因组基因预测详细的统计信息:包括contig长度、基因长度、GC含量、GC偏移、使用的RBS motif以及起始和终止密码子的使用情况。
Prodigal 不能做的: - 预测RNA基因:目前,Prodigal还不能预测RNA基因,不排除在未来的版本中增加这项功能;
- 处理含有内含子的基因:原核生物种含有内含子的基因非常罕见,所以prodiga省去了对这一部分基因的预测;
- 基因功能注释:Prodigal并不为它预测的基因提供功能注释;
- 处理移码突变(frame shifts):Prodigal不包含任何处理插入或删除的逻辑,Indel类型的测序错误将对基因预测产生有害的影响
- 病毒基因预测:尽管可以进行预测,但是目前还没有专门针对病毒基因预测的流程
Installing Prodigal
Installing on Mac OS X
# 更新homebrew
brew update
# 克隆prodigal库
brew tap hyattpd/prodigal
# 安装
brew install prodigal
Installing on Generic Unix
# 下载并解压,cd到源码目录中
# 默认安装到/usr/local/bin
make install
# 如果想更改其他安装目录
make install INSTALLDIR=/where/i/want/prodigal/
Installing on Windows
最简单的安装,点击安装包安装即可
- 版本:Prodigal V2.6.3: February, 2016
Usage
- 同时输出gff3、核酸以及蛋白序列文件
prodigal -i test.fna -f gff -o gene.gff3 -d gene.fna -a gene.faa
-i 指定待预测的基因组文件,必须指定
-f 指定输出的基因位置信息的格式:gff(default)/gbk/sqn/sco
-o 指定输出基因位置信息文件名,不写则输出到屏幕
-d 指定输出基因核酸序列文件
-a 指定输出基因蛋白序列文件
- 单个基因组和元基因组预测模式:
prodigal -i my.genome.fna –f gff -o gene.gff3 –p single
-p: 预测模式,single(default)/meta
- 改变使用的密码子表
prodigal -i my.genome.fna –f gff -o gene.gff3 –g 11
-g: 指定使用密码子表,数值1-25,默认11
11: Standard Bacteria/Archaea,标准细菌/古菌密码子表
4: Mycoplasma/Spiroplasma ,支原体和螺旋体密码子表
1-25:其他密码子表
Advice By Input Type
Finished Genomes
我们将一个完整的基因组定义为每个染色体或质粒在一个contig中,没有N′s(间隙)的基因组。
完成的基因组应该在正常模式下运行。对于确定序列的第一个和最后一个碱基不属于基因的基因组,应该考虑使用code选项。
code, codeodelosed: Closed ends. Do not allow partial genes
at edges of sequence.
如果基因组由多个染色体组成,你可以一起分析它们,也可以单独分析。只有当(1)每条染色体至少500kb,(2)你有理由相信染色体在GC含量、RBS motif用法和其他参数方面有很大的不同时,染色体才应该分开分析
质粒比较棘手,目前还不清楚最好的方法是什么。它们可以与染色体一起(这种情况下,Prodigal将在染色体和质粒一起训练),或者你可以分开分析它们,如下所述。你的决定应该再次以质粒是否与基因组的其他部分相似或不同为指导。
Draft Genomes
在大多数情况下,草稿基因组应该在正常模式下进行分析。即使平均contig长度很小(3000+bp)。或者,即使只存在一个长contig,通常也足以提供良好的训练数据。如果Prodigal在构建良好的训练集时遇到困难(由于序列在太多contigs中),它将输出如下警告:
Warning: Training sequence is highly fragmented.
You may get better results with the '-p anon' option.
默认情况下,Prodigal的参数非常适合scafflod和/或具有多个contig的FASTA序列.Prodigal允许部分基因出现N的gaps,这意味着你在一个文件中分析1000条contigs的结果,应该和分析一个由N连接的1000条contigs的scaffold得到相同的结果。此外,基因可以脱离边缘。
Prodigal可以使用-e选项以多种方式处理gaps(定义为两个或多个连续的、完全有歧义的字符的密码子)
-e, --gap_mode: Specify gap-handling behavior.
0: Partial genes run into gaps.
(Default)
1: Genes cannot run into gaps.
2: Do not treat N's as gaps.
如果你的基因组是低质量的草图,你没有一个高质量的密切相关的基因组训练,你应该在匿名模式分析序列。
提示:在有许多contigs的draft基因组时,你应该小心使用code选项,因为这将防止Prodigal预测不完整基因。类似地,如果你有一个有很多gaps的scafflod,你应该小心使用-e选项,因为你可能会失去很多不完整基因。
Metagenomes
对于宏基因组来说,最简单的方法是将所有序列放到一个FASTA文件中,并以匿名模式分析它们。这将产生合理的结果(大约有95%的效果,就像Prodigal在实际的基因组上进行训练一样)。它还具有易于并行化的优点,因为文件中的每个序列都可以独立于文件中的任何其他序列进行处理。
如果可能的话,更理想的解决方案是从样本中收集尽可能多的基因组,将每个基因组放入FASTA文件中,然后使用正常模式分析每个基因组。然后,您可以使用匿名模式分析剩余内容。
类似地,您可以使用 binning or classification程序(这些程序通常依赖于GC内容、BLAST搜索、k-mer搜索或其他信息)对序列进行binning。然后,您可以从每个bin创建多个FASTA文件,并使用普通模式分析它。
建议:不要用普通模式分析包含多个基因组序列的FASTA文件。这条规则的唯一例外是基因组密切相关(同一物种的菌株)。
上述两种解决方案都应该比匿名模式产生更好的结果,因为Prodigal能够根据序列本身进行训练而不是依赖于预设的训练文件时,它总是做得更好。但是,这些方法涉及大量的预处理工作,并且不能轻松地并行运行。最快的解决方案就是使用匿名模式。
测序错误曾经在宏基因组样本中很常见,但随着测序技术的改进,这个问题变得不那么严重了。
我们选择不花时间在Prodigal中解决此问题。 但是,如果您的读数包含许多插入/缺失,则最好使用FragGeneScan之类的程序来分析样品,该程序专门用于处理测序错误。
Alternate Genetic Codes
Prodigal支持NCBI定义的所有遗传密码。大多数细菌和古生菌使用基因密码11,它使用三个终止密码子(TAA, TGA和TAG)。有些细菌不使用TGA作为终止密码子。支原体、螺旋体和脲原体将UGA翻译成色氨酸(W)(遗传密码4),而细菌使用遗传密码25将UGA翻译成甘氨酸(G)。
默认情况下,Prodigal会尝试遗传密码11。如果平均基因长度太低,它尝试遗传密码4。如果平均基因长度仍然过低,它将恢复到遗传密码11并输出警告。如下所示
Building training set using genetic code 11...done!
Checking average training gene length...459.7, too low.
Trying genetic code 4...still bad, reverting to genetic code 11.
Redoing genome with genetic code 11...done.
Warning: Average training gene length is low (459.7).
Double check translation table or check for pseudogenes/gene decay.
Examining upstream regions and training starts...done.
Prodigal不能自动区分遗传密码4和25。在这种情况下,它可能会选择遗传密码4,您将需要使用遗传密码25手动重新运行。
自动检测是高度可靠的,不应该犯任何错误(我们测试了20,000个基因组,它没有在遗传密码的确定中犯任何错误)。但是,用户也可以使用-g选项显式地指定遗传代码。
-g, --trans_table: Specify a translation table to use.
auto: Tries 11 then 4 (Default)
11: Standard Bacteria/Archaea
4: Mycoplasma/Spiroplasma
#: Other genetic codes 1-25
这对任何使用25号基因密码的生物都是必要的,可能有些4号基因无法被自动检测到。如果您知道您的基因组的遗传密码,您可能会覆盖自动检测,并使用这个选项显式地指定它。
Organisms with Gene Decay
Gene Decay是指基因组大量序列属于非编码序列,例如假基因或者调控序列,代表物种为麻风杆菌
如果您在正常模式下运行Prodigal,并看到以下警告
Warning: Average training gene length is low (459.7).
Double check translation table or check for pseudogenes/gene decay.
那么,最终的基因预测可能“出了问题”。当其训练集中的平均基因长度小于600bp时,Prodigal打印上述警告。有些生物可能只是比一般生物的基因小。如果是这种情况,您可以忽略警告并继续正常工作
然而,有些生物可能经历了广泛的基因衰退(例如,在麻风分枝杆菌中),并且许多基因可能是基因片段,假基因,或简单的无意义(假阳性)。在这种情况下,您可能需要过滤掉许多这样的预测。
另一种可能有帮助的方法是使用训练模式对一个密切相关的基因组进行训练。例如,如果“Prodigal”对结核分枝杆菌进行了训练,而训练文件应用于麻风分枝杆菌,那么“Prodigal”预测的基因就比直接在麻风分枝杆菌上以正常模式运行时预测的基因要少得多。为了使这个过程起作用,训练的基因组必须是高质量的(而且本身没有基因组衰退),并且离你想分析的基因组不远。
Plasmids, Phages, Viruses, and Other Short Sequences
分离的短序列(<100kbp)如质粒、噬菌体和病毒一般应采用匿名模式进行分析。在质粒的情况下,你最好将质粒与染色体结合,并在正常模式下运行一切。然而,如果你相信质粒与基因组的其他部分有很大的不同,那么匿名模式是最好的选择。如果序列足够长,则应该使用普通模式。
我们从未广泛地研究过Prodigal需要多少序列才能产生良好的结果。我们通常认为20kbp-100kbp太短,无法收集足够的统计数据来很好地预测基因,但是我们没有实际数据来支持这种说法。如果您对在正常模式下运行50kbp序列感到满意,并且对所获得的结果感到满意,则可以继续这样做。尤其是起始站点需要大量数据(训练集中的100kbp起始站点可能只有约80个左右,因为Prodigal仅在较长ORF的起始位置进行训练)。我们建议使用500kbp +以获得理想的5’预测,尽管这可能在保守方面。如果您的序列在这个模糊范围内(3’预测为20kbp至100kbp,5’预测为100kbp至500kbp),则可以尝试同时运行正常模式和匿名模式,并手动检查结果是否存在差异。
Prodigal不包含处理病毒的特殊程序。因此,它无法处理某些有时在病毒中发生的现象,例如翻译移码。通常应按上述方法分析病毒,以匿名模式分析短基因组,以正常模式分析较长基因组。
Understanding the Prodigal Output
Gene Coordinates
gene coordinates文件列出了每个基因的位置以及一些额外的评分信息。默认情况下,Prodigal生成一个类似genbank的特征表;但是,用户可以通过-f选项指定其他一些输出类型
-f, --output_format: Specify output format.
gbk: Genbank-like format (Default)
gff: GFF format
sqn: Sequin feature table format
sco: Simple coordinate output
gff参数产生通用特征格式版本3输出。 尽管用户仍然必须填写产品信息以具有“可提交”的Sequin文件,但sqn参数生成Sequin Feature Table格式。 sco参数生成简单坐标输出,如果用户只需要基因坐标而没有其他要求,则适用。
对于FASTA输入文件中的每个单独序列,Prodigal会生成一个标头,其中包含以分号分隔的字符串,其中包含有关该序列及其分析方式的信息(以名称=值对的形式)。 在Genbank格式中,将其放置在“ DEFINITION”行上。 在GFF3中,此信息分为两个注释行。
Sample Genbank header:
DEFINITION seqnum=1;seqlen=4639675;seqhdr="NC_000913 # Escherichia coli str. K-12 substr. MG1655, complete
genome.";version=Prodigal.v2.60;run_type=Single;model="Ab initio";gc_cont=50.79;transl_table=11;uses_sd=1
Sample GFF3 header:
##gff-version 3
# Sequence Data: seqnum=1;seqlen=4639675;seqhdr="NC_000913 # Escherichia coli str. K-12 substr. MG1655, com
plete genome."
# Model Data: version=Prodigal.v3.0.0-devel.1.0;run_type=Normal;model="Ab initio";gc_cont=50.79;transl_tabl
e=11;uses_sd=1
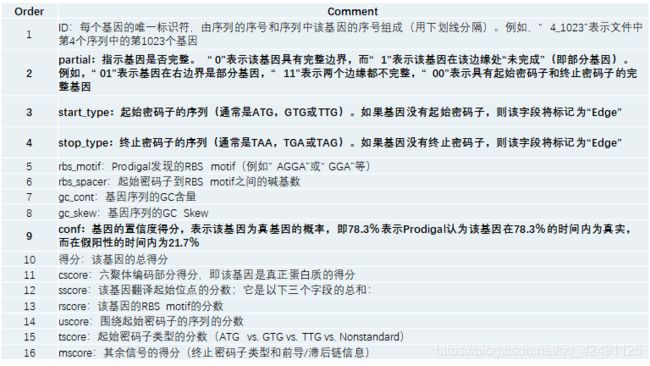
每一列的意义:
seqnum: An ordinal ID for this sequence, beginning at 1.
seqlen: Number of bases in the sequence.
seqhdr: The entire FASTA header line.
version: Version of Prodigal used to analyze this sequence.
run_type: "Ab initio" for normal mode, "Anonymous" for anonymous mode.
model (Anonymous mode only): Information about the preset training file used to analyze the sequence.
gc_cont: % GC content of the sequence.
transl_table: The genetic code used to analyze the sequence.
uses_sd: Set to 1 if Prodigal used its default RBS finder, 0 if it scanned for other motifs.
除了链(由GFF中的“ +/-”标签或Genbank格式中指示反向链的“ complement”关键字指定)和基因边界外,Prodigal还会生成以分号分隔的名称-值对字符串( (以name = value的形式)以及有关每个基因的得分和统计信息。
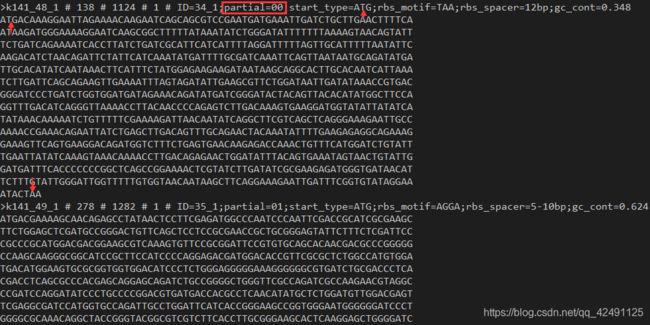
In Genbank format, this is placed on a “note” line, like so:
CDS 2655..2882
/note="ID=1_3;partial=00;start_type=ATG;stop_type=TAA;rbs_motif=AGxAGG/AGGxGG;rbs_spac
er=5-10bp;gc_cont=0.241;conf=100.00;score=44.71;cscore=32.81;sscore=11.90;rscore=9.40;uscore=-1.05;tscore=3
.55;"
In GFF3 format, the string is placed in the last field, so the gene occupies only one line instead of two, like so:
NC_000913 Prodigal_v3.0.0-devel.1.0 CDS 337 2799 338.7 + 0 ID=1_2;part
ial=00;start_type=ATG;stop_type=TGA;rbs_motif=GGAG/GAGG;rbs_spacer=5-10bp;gc_cont=0.531;conf=99.99;score=33
8.70;cscore=322.16;sscore=16.54;rscore=11.24;uscore=1.35;tscore=3.95;
GFF3格式要求在第一个字段中有一个“id”。FASTA标题的第一个词和使用,作为其id。这个id是不能保证是唯一的(各种标题的第一个词的文件可以是相同的),所以我们建议用户依靠semicolon-delimited字符串中的“id”字段。
GFF3格式:
ID: A unique identifier for each gene, consisting of the ordinal ID of the sequence and an ordinal ID of that gene within the sequence (separated by an underscore). For example, "4_1023" indicates the 1023rd gene in the 4th sequence in the file.
partial: An indicator of if a gene runs off the edge of a sequence or into a gap. A "0" indicates the gene has a true boundary (a start or a stop), whereas a "1" indicates the gene is "unfinished" at that edge (i.e. a partial gene). For example, "01" means a gene is partial at the right boundary, "11" indicates both edges are incomplete, and "00" indicates a complete gene with a start and stop codon.
start_type: The sequence of the start codon (usually ATG, GTG, or TTG). If the gene has no start codon, this field will be labeled "Edge".
stop_type: The sequence of the stop codon (usually TAA, TGA, or TAG). If the gene has no stop codon, this field will be labeled "Edge".
rbs_motif: The RBS motif found by Prodigal (e.g. "AGGA" or "GGA", etc.)
rbs_spacer: The number of bases between the start codon and the observed motif.
gc_cont: The GC content of the gene sequence.
gc_skew: The GC skew of the gene sequence.
conf: A confidence score for this gene, representing the probability that this gene is real, i.e. 78.3% means Prodigal believes that gene is real 78.3% of the time and a false positive 21.7% of the time.
score: The total score for this gene.
cscore: The hexamer coding portion of the score, i.e. how much this gene looks like a true protein.
sscore: A score for the translation initiation site for this gene; it is the sum of the following three fields.
rscore: A score for the RBS motif of this gene.
uscore: A score for the sequence surrounding the start codon.
tscore: A score for the start codon type (ATG vs. GTG vs. TTG vs. Nonstandard).
mscore: A score for the remaining signals (stop codon type and leading/lagging strand information).
Protein Translations
蛋白质翻译文件由所有序列中的所有蛋白质以多种FASTA格式组成。FASTA标头以一个文本id开始,该id由原始FASTA序列标头的第一个单词、下划线和蛋白质的序号id组成。这个文本id不能保证是唯一的(它取决于用户提供的FASTA报头),这就是为什么我们建议在最后一个分号分隔的字符串中使用“id”字段。
An example header for the 4th protein in the E. coli genome with id NC_000913:
>NC_000913_4 # 3734 # 5020 # 1 # ID=1_4;partial=00;start_type=ATG;rbs_motif=GGA/GAG/AGG;rbs_spacer=5-10bp;g
c_cont=0.528
标头中的后三个字段由“#”符号分隔,分别是基因组中最左边的坐标,最右边的坐标和链(正向链基因为1,反向链基因为-1)。 坐标信息后是一个用分号分隔的字符串,与基因坐标文件中描述的字符串相同(请参见此处的列表以获取字段定义),仅使用以下字段:ID,partial,start_type,stop_type,rbs_motif,rbs_spacer,gc_cont, 和gc_skew和conf。 除conf字段外,标头不包含有关该基因的任何评分信息。
Nucleotide Sequences
核苷酸序列文件按照蛋白质翻译部分中描述的相同规则和约定产生多个FASTA输出。唯一值得注意的是,Prodigal使用DNA字母表来产生这些序列,而不是mRNA(所以你会在输出中看到’T’,而不是’U’).
Starts File
Summary Statistics
prodigal -i my.metagenome.fna -o my.genes -a my.proteins.faa -p meta
nohup time prodigal -a HTR8.faa -d HTR8.fna -f gff -o HTR8.gff -p meta -i ../HTR8_Megahit.fa &>HTR8.log &
-a: 输出选中文件的蛋白翻译结果.
code: 不允许注释的基因超出基因组序列的边缘.
-d: 将基因序列输出到该文件
-f: 选择输出的注释文件格式 (gbk, gff, or sco). Default is gbk.
-g: 指定翻译密码子 (default 11).
-h: 打印帮助菜单并退出.
-i: 指定输入的fasta和genbank文件 (default reads from stdin).
-m: 将包含N的序列过滤掉; 不在该区段注释文件.
-o: 指定输出的文件 (default writes to stdout).
-p: 选择注释程序 (单基因组 or 宏基因组). Default is single.
-q: 静默模式,不在屏幕输出任何错误信息 (suppress normal stderr output).
-s: 输出所有潜在的基因及其得分
-v: 打印版本信息并退出.
输出结果
ID: 每个基因的唯一ID,包含每条序列的序号ID。以及原始的基因ID,以及序列的次序ID(用下划线隔开).
例如, "4_1023"表示第1023个基因在文件的第四条序列中。
partial:表示一个基因是否达到序列的边界,或者引入了Gap缺失。 “0” 表示基因有一个真正的边界
(起始 or 终止),1表示基因在边界上未结束的,也就是部分基因。例如, "01"表示基因在右边界是缺失的,
“11” 表示基因在两端都是缺失的, 以及 “00"表示一个具有起始和终止密码子的完整基因。
start_type: 序列起始密码子的类别 ( ATG, GTG, or TTG). 如果该基因没有起始密码子,这一域写作"Edge”.
stop_type:序列终止密码子的类别(通常是TAA, TGA, or TAG).如果该基因没有终止密码子,这一域叫做 “Edge”.
rbs_motif: Prodigal发现的核糖体结合的motif (e.g. “AGGA” or “GGA”, etc.)
rbs_spacer: 起始密码子和rbs motif之间的碱基数目
gc_cont:基因序列的GC含量.
gc_skew:基因序列的GC偏度.
conf: 该基因的置信得分,代表该基因真正为基因的概率.
score: 该基因的所有得分.
sscore: 该基因的翻译起始位点得分; 是以下三部分的得分之和.
rscore: 该基因的RBS motif 得分.
uscore: 围绕着起始密码子的序列得分.
tscore: 起始密码子类型的得分 (ATG vs. GTG vs. TTG vs. Nonstandard).
mscore: 剩余的信号得分 (stop codon type and leading/lagging strand information).
早期版本:Gene Prediction Modes
三种预测模式:Normal Mode/ Anonymous Mode/ Training Mode
这些方法中哪一种适合您的序列取决于您要分析的数据集类型。当你有足够的数据可以训练时,你就应该使用普通模式(100k bp+用于3’预测,500k bp+用于5’预测,这是安全的数字,尽管你可以使用更少的数据)。匿名模式应用于元基因组数据集,或用于太短而不能提供良好训练数据的序列。
不同模式的特点:
-p, --mode: Specify mode (normal, train, or anon).
normal: Single genome, any number of
sequences. (Default)
train: Do only training. Input
should be multiple FASTA of
one or more closely
related genomes.
anon: Anonymous sequences, analyze
using preset training files,
ideal for metagenomic data
or single short sequences.
meta: (Deprecated) Same as anon.
single: (Deprecated) Same as normal.
Normal Mode
Prodigal对输入序列,进行学习,了解其特性,然后根据这些特性预测基因。
适用于完成的基因组,质量合格的草图基因组和大型病毒。
在单个或多个FASTA序列上运行normal mode
prodigal -i my.genome.fna -o gene.coords.gbk -a protein.translations.faa
-i 输入文件, 可以是single/multiple FASTA, Genbank, or EMBL format,推荐FASTA
-o 输出文件(基因坐标)
-a 输出蛋白翻译文件
在没有指定输入和输出的时候,Prodigal从标准输入并标准输出:
prodigal < my.genome.fna > gene.coords.gbk
完整的参数:
-a: Write protein translations to the selected file.
code: Closed ends. Do not allow genes to run off edges.
-d: Write nucleotide sequences of genes to the selected file.
-f: Select output format (gbk, gff, or sco). Default is gbk.
-g: Specify a translation table to use (default 11).
-h: Print help menu and exit.
-i: Specify FASTA/Genbank input file (default reads from stdin).
-m: Treat runs of N as masked sequence; don't build genes across them.
-n: Bypass Shine-Dalgarno trainer and force a full motif scan.
-o: Specify output file (default writes to stdout).
-p: Select procedure (single or meta). Default is single.
-q: Run quietly (suppress normal stderr output).
-s: Write all potential genes (with scores) to the selected file.
-t: Write a training file (if none exists); otherwise, read and use
the specified training file.
-v: Print version number and exit.
Anonymous Mode
Prodigal将预先计算的训练文件应用于提供的输入序列,并根据最佳结果预测基因.
匿名模式应用于元基因组,低质量的草图基因组,小型病毒和小型质粒。
运行anonymous mode,只需添加 ‘-p anon’,例如:
prodigal -i metagenome.fna -o coords.gbk -a proteins.faa -p anon
Training Mode
它的工作方式与普通模式类似,但Prodigal会保存训练文件以备将来使用.
训练模式(第三个选项)的工作方式与普通模式类似,但是会输出一个训练文件,可用于以后的分析。这主要在您希望按照与您要分析的序列不同的序列进行训练时很有用。
Prodigal还有一个训练模式,可以编写训练数据文件供以后使用。使用此模式的主要原因是,如果您希望对一个输入序列进行训练,然后对另一个输入序列进行分析。比如,在基因组1上训练,在基因组2上运行,就可以了.
prodigal -i genome1.fna -p train -t genome1.trn
# This writes a training data file to genome1.trn, as specified by the -t option.
-t, --training_file: Specify the training file location. In
train mode, writes the training file.
In normal mode, reads the training file.
# To read in the genome 1 training file and use it to analyze genome 2, one would do:
prodigal -i genome2.fna -t genome1.trn -o genome2.gbk -a genome2.faa
耗时
在500G机器上使用30核,meta模式计算27801017(27M)contig(>1k)耗时75h
统计全长基因
(cat) [yutao@globin ColdSeepNonRedundant_GeneSets]$ awk '$0~/>/ && $9~/partial=00/{print $5-$3}' ColdSeep_Gene_ID95_Cov0.8_rep_seq.fasta|awk '{sum+=$1}END{print NR,sum/NR}'
参考
prodigal
prodigal wiki