技术干货 | MindSpore AI科学计算系列(四):AlphaFold2分析
背景
蛋白质折叠这个被列为“21世纪的生物物理学”的重要课题之一,已经困扰了科学家们近半个世纪,好消息是在两年一届的CASP比赛中,2020年谷歌deepmind团队的Alphafold2凭借其接近90分的成绩取得CASP14比赛中蛋白质3D结构预测的榜首,这一成就被Nature等杂志喻为前所未有的进步。
近期AlphaFold2连续两登Nature着实在又生物和科研圈火了一把,首先是在7月15日宣布开源,
https://github.com/deepmind/alphafold
并将长达六十多页的代码解析及数据集对外公开,我们团队也第一时间对代码进行了复现,并重跑了CASP14的比赛数据集,发现结果确如人所愿,大部分的结果均可达到将近90分(TM-score)的高分,然而一些序列不太长的蛋白质序列的结果反而差强人意,其原因我在后文会进一步介绍。紧随其后一周之后的7月22日,谷歌再度Nature发声,宣布将会把覆盖人类98.5%的蛋白质预测结果对外公开,其中58%达到了可信水平,35.7%达到了高置信度,要知道之前科学家经过几十年的努力靠着实验观测手段才覆盖了人类蛋白质序列中17%的蛋白质。
那么什么是蛋白质折叠问题呢
引用维基百科的描述如下:蛋白质折叠(英语:Protein folding)是蛋白质获得其功能性结构和构象的物理过程。通过这一物理过程,蛋白质从无规则卷曲折叠成特定的功能性三维结构。在从mRNA序列翻译成线性的氨基酸链时,蛋白质都是以去折叠多肽或无规则卷曲的形式存在。
简单来说蛋白质是由氨基酸序列组成的,人体内常见氨基酸种类为20种,这20种氨基酸的排列组合形成特定的氨基酸链时,经过mRNA翻译形成氨基酸的肽链,随后由于氨基酸之间在分子力的作用下扭曲旋转而形成三维空间结构的过程即是蛋白质折叠过程,而最终形成的这个空间结构决定了蛋白质的功能特性。因此如果能快速知道一种蛋白质的空间结构,那么科学家就可以了解其生物功能特性,如何通过蛋白质的一级氨基酸序列获得其生理形态的三维空间结构即是蛋白质折叠问题。

表1:常见20种氨基酸
为什么说蛋白质折叠问题困扰人类近半个世纪呢
想要得到蛋白质的空间结构传统的观测手段为通过冷冻电镜,x射线晶体衍射,二维核磁共振等手段。对于X射线晶体衍射和二维核磁共振,需要经过基因表达,蛋白质提取和纯化,蛋白质结晶等过程,其中仅仅获取纯度较高的蛋白质晶体这一步就非常困难,不仅要考虑温度还要考虑溶剂环境等的影响,实验周期可达数月之久,近年来出现的冷冻电镜虽观测精度高,但价格昂贵(单价数千万人民币),对操作手段,观测一个蛋白质就需几十万及以上的的成本。
另外1958年F.H.C. 克里克提出了生物学中著名的中心法则:DNA-RNA-蛋白质,并指出蛋白质是由DNA转录为RNA,再由RNA翻译成一个个氨基酸组合而成的,因此蛋白质的一级氨基酸序列信息应该包含了蛋白质三维空间结构的必要重要信息。从此越来越多的人开始尝试通过科学计算的方法从蛋白质一维氨基酸序列直接推导出其三维构想,但因蛋白质的每个氨基酸的侧链,甚至氨基酸与氨基酸之间形成的肽键均可以旋转,其每个原子位置均需要确定,因此整个蛋白质的空间构想种类可达10^300之大,这可比宇宙中所有原子的数量还大,由此出现了CASP(Critical Assessment of protein Structure Prediction)比赛,用于激励大家参与解决蛋白质折叠问题,该赛事成立于1994年,每两年举办一次,也是计算生物学领域中最权威和最富盛名的比赛,最新2020年的比赛为CASP14,alphafold2即在该比赛中拿到了结构预测的第一名。
解决蛋白质折叠问题有什么意义
解决蛋白质折叠问题后可从蛋白质的一维序列获取其三维结构,这对新药研发,蛋白质设计有着至关重要的作用。
如人体内的抗体蛋白,就是通过其特殊的“Y”型结构和特定的病毒或细菌结合从而消灭他们,再比如人体内的胶原蛋白,其结构如绳索一般,具有很强的韧性,可以传递张力,广泛存在于软骨,韧带等,了解蛋白质的空间结构后就可以更好的了解其活性中心,配体靶点等,甚至可以改造蛋白质从而让其满足特性的功能需求。
传统的药物设计一般是通过筛选大量的天然化合物,寻找易与目标蛋白质分子紧密结合,容易合成且没有毒副作用来完成的,因此研发周期较长,费用较高,而了解蛋白质的空间结构后可以减少这种寻找药物的盲目性,缩短研发周期和降低成本。
AlphaFold2算法相关
整个算法框架如图所示为end2end结构,可以大体分为三个部分,分别是数据处理,网络和结构预测,总输入为单个蛋白质序列。数据处理部分集成了生成MSA和查找templates(模板,包含了MSA中与源序列相似的已知蛋白质结构信息)的部分,另外还引入了pairing信息(包含每个残基-残基对儿的特征。简单来说每个残基之间都有一个隐性的状态来描述其之间的关系,这个信息即被文章里称为pairwise features)。
MSA, pairing, templates 被输入到Evoformer网络中,其中pair 信息和MSA信息相互迭代更新,并输入到结构预测模块Structure module,最终得到3D结构信息。
Evoformer 模块

Evoformer模块的输入如上图所示,因alphafold2选取的MSA长度为128,但很多蛋白质的MSA包含了最多5000条蛋白质序列,因此这里除了随机选取128条作为输入外,还将剩余的MSA序列对选取的作中心聚类,然后作为extra-msa信息, template信息与MSA和pair进行简单的相加与concat之后再加入extra-msa信息作为整个Evoformer的输入部分。

在网络架构中接收前面提取的MSA和pair表征后,不仅有横向的attention对当前氨基酸序列的所谓位置进行attention计算,还有纵向的attention去获取其他MSA序列相同位置上氨基酸的突变与稳定性信息。MSA经过横纵attention之后再经过一个transition层,然后通过计算外积并取平均来计算pairwise features。对于pairwise features的更新采用的是三角法则原理,原因是pairwise features信息记录了残基之间的两两距离关系,因距离关系不是自由的,应该满足三角不等式,即:相邻的三个边应该满足两边之和大于等于第三边,所以对于每个边,即pairwise features里的每个pair信息都会接收和其相关的可组成三角形的任意两个边来进行更新。
Structure module 部分
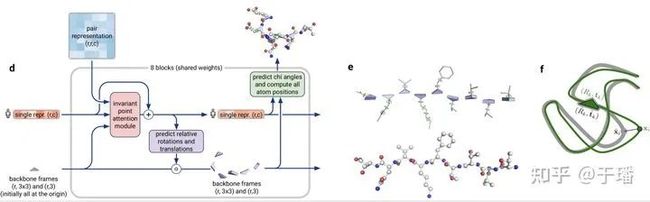
接着就来到了结构更新模块,该模块以经过Evoformer 网络层后得到的pair features信息,原始的序列信息(MSA中只保留目标氨基酸序列,丢弃剩余MSA作为single repr)以及将所有残基位置的初始化空间结构信息作为输入,首先经过IPA(invariant point attention)模块更新序列表征信息,更新后的single repr被映射到主链上通过欧几里得变换并更新坐标信息,在通过计算主链以及侧链的旋转角度信息,最终更新得到预测的全原子坐标,感兴趣的可以查看alphafold2的suppl文献。
了解了alphafold2的整体流程之后,可以发现MSA以及template的信息对于最终的预测结果起到非常重要的作用,从预测结果来看,对于同源已知蛋白质数量越多的目标序列,其预测结果准确率越高,而对于同源蛋白质数量很少的那些预测结果则较差,如我们使用AlphaFold2预测了CASP14中的T1043号蛋白,虽然其氨基酸序列长度仅为148(相比CASP14其他蛋白较小),但是预测结果却比较差(如图所示,蓝色为预测值,红色为真实值),TM-score仅为20多,原因就是该序列的同源模板数量仅为个位数。

总结一下
1. Alphafold2广泛使用了transformer结构,不管是MSA还是残基-残基对的信息更新都使用了attention机制,结构模块的更新使用了三角法则,简化了计算的复杂度,准确率也提高了不少。
2. 整个模型的Evoformer和structure module部分都使用了recycling,即将输出重新加入到输入在重复refinement,进行信息的精炼。
3. 对于训练部分,先进行了预训练,然后把MSA中没有标签的序列预测出三维结构,再将这些训练结果置信度较高的结构保存下来重新进行训练
4. 对msa还做了masked attention自监督训练
然而我们发现在推理的过程中使用到的都是现有的蛋白库,对于那些MSA序列长度还可以,但是模板较少的目标序列而言,其预测精度较低,那是不是可以在推理的过程中也把MSA中没有结构信息的序列进行预测,再把结果保留下来当做template来做预测呢,这个感兴趣的同学也可以一起思考下。
参考资料
1. https://predictioncenter.org/index.cgi
2. https://alphafold.ebi.ac.uk/
3. Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021). https://doi.org/10.1038/s41586-021-03819-2
4. https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-021-03819-2/MediaObjects/41586_2021_3819_MOESM1_ESM.pdf
5. https://zh.wikipedia.org/wiki/蛋白质折叠