2023-11-28-直播单细胞图表美化-seurat数据结构 featureplot dotplot vlnplot










单细胞常见的可视化方式有DimPlot,FeaturePlot ,DotPlot ,VlnPlot 和 DoHeatmap几种 ,Seurat中均可以很简单的实现,但是文献中的图大多会精美很多。
之前 跟SCI学umap图| ggplot2 绘制umap图,坐标位置 ,颜色 ,大小还不是你说了算 介绍过DimPlot的一些调整方法。
本文介绍FeaturePlot的美化方式,包含以下几个方面 :
(1)调整点的颜色 ,大小
(2)展示基因共表达情况(点图,密度图)
(3)优化Seurat分组展示
(4)ggplot2修改theme ,lengend等
(5)批量绘制
一 载入R包,数据
仍然使用之前注释过的sce.anno.RData数据 ,后台回复 anno 即可获取
library(Seurat)
library(tidyverse)
library(scCustomize) # 需要Seurat版本4.3.0
library(viridis)
library(RColorBrewer)
library(gridExtra)
load("sce.anno.RData")
head(sce2,2)
这里额外安装scCustomize包,该R包对上面提到的Seurat 常用绘图函数进行了一些优化,但是需要Seurat版本4.3.0 以上。
二 FeaturePlot 相关
1,调整FeaturePlot颜色,大小
(1)Seurat 修改
有以下几种方式,可以使用FeaturePlot 内置的cols参数进行修改(p2 , p3),也可以使用ggplot2的方式 添加scale 进行修改(p4)
p1 <- FeaturePlot(object = sce2, features = "CD3D")
p2 <- FeaturePlot(sce2, "CD3D", cols = c("#F0F921FF", "#7301A8FF"))
p3 <- FeaturePlot(sce2, "CD3D", cols = brewer.pal(10, name = "RdBu"))
p4 <- FeaturePlot(object = sce2, features = "CD3D") +
scale_colour_gradientn(colours = rev(brewer.pal(n = 10, name = "RdBu")))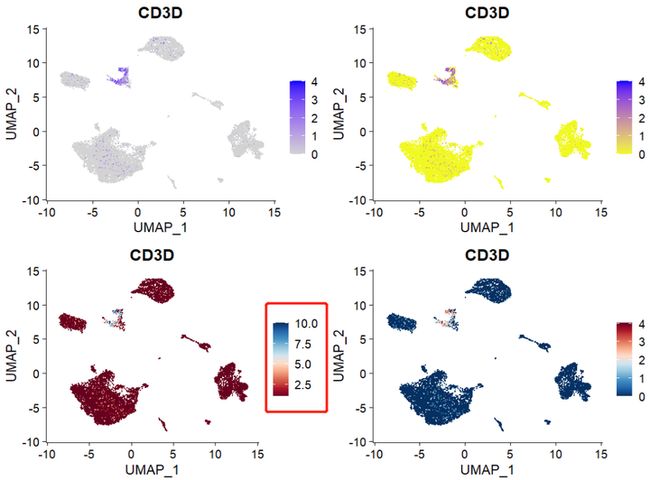
注意左下p3 ,legend是有问题的,会随col参数中brewer.pal(10, name = "RdBu")中的10的数值而变动。
修改大小的话很简单,直接调整 pt.size = 1 即可,此处不做演示。
(2)scCustomize 修改
p11 <- FeaturePlot_scCustom(seurat_object = sce2, features = "CD3D")
p22 <- FeaturePlot_scCustom(seurat_object = sce2, features = "CD3D", colors_use = brewer.pal(11, name = "RdBu"),order = T)
p11 + p22
这里cols参数是没有问题的。
2 ,多基因共“表达”
单个基因就按照上面的方法直接绘制即可,如果想同时显示2个基因呢?
(1)Seurat 中提供了 blend = TRUE 函数,来可视化两个基因的共表达情况
FeaturePlot(sce2, features = c("MS4A1", "CD79A"), blend = TRUE)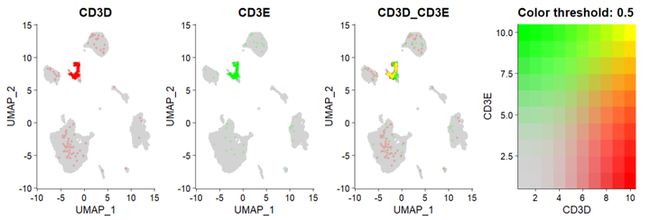
注意blend = TRUE函数只能适用于2个基因,多个基因会报错 。
如果想实现多个基因的话,将目标基因和UMAP 的坐标提取出来使用ggplot2绘制即可 或者 使用scCustomize 包中的多基因联合密度图 ,如下。
(2)scCustomize 多基因联合密度图
密度图是通过Nebulosa包实现的,因此需要先安装Nebulosa 包 。然后用Plot_Density_Joint_Only()函数即可以同时绘制多个基因的联合密度图 ,可以不限于2个基因 。
BiocManager::install("Nebulosa")
#单基因
p000 <- Plot_Density_Custom(seurat_object = sce2, features = "CD3D")
#双基因
p111 <- Plot_Density_Joint_Only(seurat_object = sce2,
features = c("CD3D", "CD3E"))
#多基因
p222 <- Plot_Density_Joint_Only(seurat_object = sce2,
features = c("CD3D", "CD3E","CD79A"),
custom_palette = BlueAndRed())
p000 + p111 + p222
可以通过custom_palette 函数调整颜色,选择较少 。
除了展示共表达外,还可展示目标celltype的几个marker来辅助细胞类型鉴定。
3 , 分组相关
很多时候会需要分样本/分组展示重点基因来进行表达的比较,
(1)Seurat有 split.by 函数 ,虽然可以设置ncol,但是没有效果,如图所示,
sce2sub <- subset(sce2 ,group == "PT")
FeaturePlot(sce2sub, "CD3D", cols = brewer.pal(11, name = "RdBu"),
pt.size = 1,
split.by = "sample" ,ncol = 4)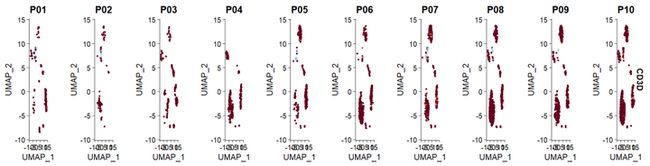
(2)scCustomize 中FeaturePlot_scCustom函数 ,算是修正了这个小bug
FeaturePlot_scCustom(seurat_object = sce2, features = "CD3D", split.by = "orig.ident",
num_columns = 4)
4 ,ggplot2 修改theme / legend 相关
类似前面使用ggplot2的scale修改颜色,当然也可以修改theme等一系列
FeaturePlot(object = sce2, features = "CD3D",pt.size = 1,order = T) +
scale_colour_gradientn(colours = rev(brewer.pal(n = 10, name = "RdBu"))) +
DarkTheme() +
theme(text=element_text(size=14))+
theme(text=element_text(face = "bold"))+
theme(legend.text=element_text(size=8))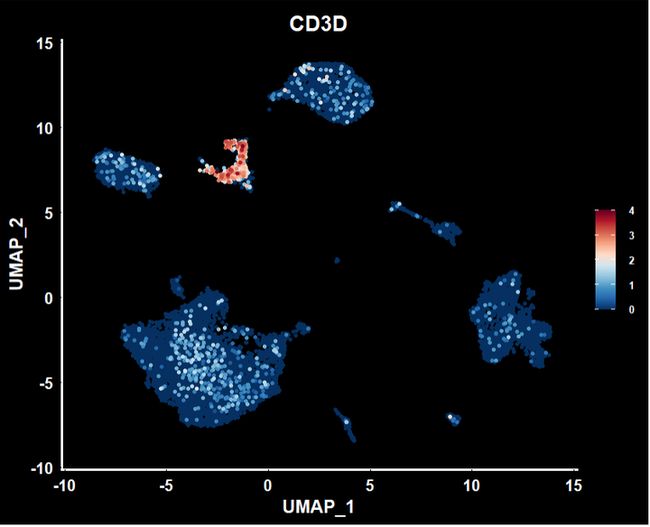
此处简单的示例,更多的参考ggplot2 | 关于标题,坐标轴和图例的细节修改,你可能想了解 , ggplot2|theme主题设置,详解绘图优化-“精雕细琢” ,和ggplot2 |legend参数设置,图形精雕细琢
5 批量绘制
当有多个基因需要绘制时候,需要批量绘制 。
(1)features 可以接受向量,因此可以直接完成
marker_sign <- c("CD3E", 'CD3D', 'EPCAM', 'CD4', 'CD8A','SPP1', 'CD19', 'COL1A1', 'IGLC1')
FeaturePlot(sce2,features = marker_sign)
(2)grid.arrange 方式绘制
grid.arrange接受的是list ,可以通过layout_matrix 调整布局 。当然也可以最开始调整好基因在向量中的顺序,Seurat的结果是一样的 。
intersect_tls <- intersect(marker_sign,rownames(sce2))
plot_list <- lapply(intersect_tls,function(x){
plot_list <- FeaturePlot(sce2,
features = x)
})
#设置布局
lay <- rbind(c(1,2,3),
c(4,5,6),
c(7,8,9))
grid.arrange(grobs = plot_list, layout_matrix = lay)因为单细胞的FeaturePlot的都是样的,看不出来grid.arrange的优势,后面会介绍空转中使用该函数通过布局 和 选择展示的图片 来绘制CNS级别的主图。
1. tsne展示marker基因
FeaturePlot(mye.seu,features = "CCR7",reduction = "tsne",pt.size = 1)+
scale_x_continuous("")+scale_y_continuous("")+
theme_bw()+ #改变ggplot2的主题
theme( #进一步修改主题
panel.grid.major = element_blank(),panel.grid.minor = element_blank(), #去掉背景线
axis.ticks = element_blank(),axis.text = element_blank(), #去掉坐标轴刻度和数字
legend.position = "none", #去掉图例
plot.title = element_text(hjust = 0.5,size=14) #改变标题位置和字体大小
)
ggsave("CCR7.pdf",device = "pdf",width = 10,height = 10.5,units = "cm") 
另一种方法就是把tsne的坐标和基因的表达值提取出来,用ggplot2画,其实不是很必要,因为FeaturePlot也是基于ggplot2的,我还是演示一下
mat1=as.data.frame(mye.seu[["RNA"]]@data["CCR7",])
colnames(mat1)="exp"
mat2=Embeddings(mye.seu,"tsne")
mat3=merge(mat2,mat1,by="row.names")
#数据格式如下:
> head(mat3)
Row.names tSNE_1 tSNE_2 exp
1 N01_AAACGGGCATTTCAGG_1 5.098727 32.748145 0.000
2 N01_AAAGATGCAATGTAAG_1 -24.394040 26.176422 0.000
3 N01_AACTCAGGTAATAGCA_1 11.856730 8.086553 0.000
4 N01_AACTCAGGTCTTCGTC_1 10.421878 12.660407 0.000
5 N01_AACTTTCAGGCCATAG_1 33.555756 -10.437406 1.606
6 N01_AAGACCTTCGAATGGG_1 -23.976967 11.897753 0.738
mat3%>%ggplot(aes(tSNE_1,tSNE_2))+geom_point(aes(color=exp))+
scale_color_gradient(low = "grey",high = "purple")+theme_bw()
ggsave("CCR7.2.pdf",device = "pdf",width = 13.5,height = 12,units = "cm")
用ggplot2的好处就是图形修改很方便,毕竟ggplot2大家都很熟悉
2. 热图展示marker基因
画图前,需要给每个细胞一个身份,因为我们跳过了聚类这一步,此处需要手动赋值
Idents(mye.seu)="celltype"
library(xlsx)
markerdf1=read.xlsx("ref_marker.xlsx",sheetIndex = 1)
markerdf1$gene=as.character(markerdf1$gene)
# 这个表格整理自原文的附表,选了53个基因
#数据格式
# > head(markerdf1)
# gene celltype
# 1 S100B DC2(CD1C+)
# 2 HLA-DQB2 DC2(CD1C+)
# 3 FCER1A DC2(CD1C+)
# 4 CD1A DC2(CD1C+)
# 5 PKIB DC2(CD1C+)
# 6 NDRG2 DC2(CD1C+)
DoHeatmap(mye.seu,features = markerdf1$gene,label = F,slot = "scale.data")
ggsave("heatmap.pdf",device = "pdf",width = 23,height = 16,units = "cm")label = F不在热图的上方标注细胞类型,
slot = "scale.data"使用scale之后的矩阵画图,默认就是这个
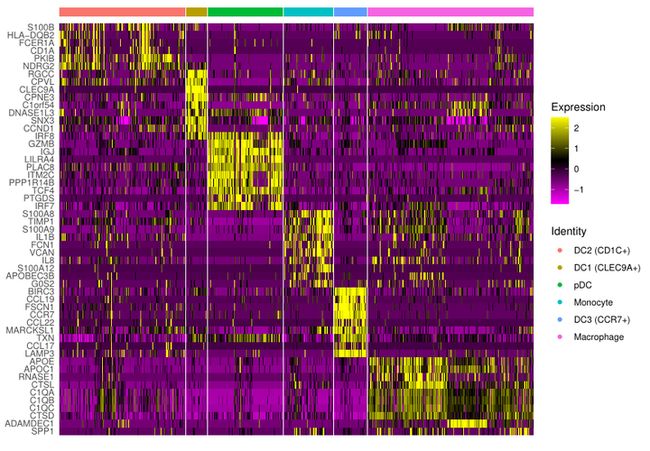
接下来用pheatmap画,在布局上可以自由发挥
library(pheatmap)
[email protected][,c("CB","celltype")]
colanno=colanno%>%arrange(celltype)
rownames(colanno)=colanno$CB
colanno$CB=NULL
colanno$celltype=factor(colanno$celltype,levels = unique(colanno$celltype))先对细胞进行排序,按照celltype的顺序,然后对基因排序
rowanno=markerdf1
rowanno=rowanno%>%arrange(celltype)提取scale矩阵的行列时,按照上面的顺序
mat4=mye.seu[["RNA"]]@scale.data[rowanno$gene,rownames(colanno)]
mat4[mat4>=2.5]=2.5
mat4[mat4 < (-1.5)]= -1.5 #小于负数时,加括号!下面就是绘图代码了,我加了分界线,使其看上去更有区分度
pheatmap(mat4,cluster_rows = F,cluster_cols = F,
show_colnames = F,
annotation_col = colanno,
gaps_row=as.numeric(cumsum(table(rowanno$celltype))[-6]),
gaps_col=as.numeric(cumsum(table(colanno$celltype))[-6]),
filename="heatmap.2.pdf",width=11,height = 7
)