一个转录组上游分析流程 | Hisat2-Stringtie
本期的教程代码(部分)
#!/bin/bash
#
# 使用fastq-dump解压sra数据
# 本数据集为双端数据
# 解压格式为fq.gz
for i in SRR6929571 SRR6929572 SRR6929573 SRR6929574 SRR6929577 SRR6929578;
do
pfastq-dump --split-files --threads 20 --gzip -s 00_RawData/${i}.sra --outdir 00_RawData/
## 质控
fastp -i 00_RawData/${i}_1.fastq.gz -o 01_CleanReads/${i}_1.clean.fq.gz -I 00_RawData/${i}_2.fastq.gz -O 01_CleanReads/${i}_2.clean.fq.gz -q 20 -z 4 -w 20 -h 01_CleanReads/html/${i}.html
## fastqc评估
fastqc -q -t 30 -o 01_CleanReads/fastqc/ 01_CleanReads/${i}_*.fq.gz
## 根据的信息,修改下面脚本
#mkdir 03_MappedFile/Hisat2_Mapped
#mkdir 03_MappedFile/Hisat2_Mapped/summary/
#mkdir 03_MappedFile/Hisat2_Mapped/Unmapped_reads
....
....
....
....
done
以下为获得.sort.bam文件后进行运行。
本教程详细教程
https://mp.weixin.qq.com/s/A4cFpkrKGqPeESVQl69jcA
# 合并gtf文件
ls 04_Result/Stringtie/*.gtf > 04_Result/Stringtie/mergelist.txt
stringtie --merge -F 0 -T 0 -G 02_Geneome_index/ITAG4.1_gene_models.gtf -o 04_Result/Stringtie/gffcompare/stringtie_merged.gtf 04_Result/Stringtie/mergelist.txt
## gffcomapre注释
gffcompare -r 02_Geneome_index/ITAG4.1_gene_models.gtf -G -o 04_Result/Stringtie/gffcompare/merged 04_Result/Stringtie/gffcompare/stringtie_merged.gtf
##
## 计算FPKM
mkdir 04_Result/Stringtie/featureCounts
featureCounts -T 20 -p -t exon -g transcript_id -a 04_Result/Stringtie/gffcompare/stringtie_merged.gtf -o 04_Result/Stringtie/featureCounts/All.transcript.count.txt 03_MappedFile/Hisat2_Mapped/*.sort.bam
###
## Count to FPKM
cat 04_Result/Stringtie/featureCounts/All.transcript.count.txt | cut -f 1,6-13 > 04_Result/Stringtie/featureCounts/01.all.count.txt
perl CountToFPKM.pl 04_Result/Stringtie/featureCounts/01.all.count.txt > 04_Result/Stringtie/featureCounts/02.all.FPKM.txt
一、写在前面
今天分享一个转录组上游分析的流程(Hisat2-Stringtie-Count),此流程的操作依旧是非常简单的。我们的流程主要使用软件的安装、数据下载、过滤、比对、Count、Count To FPKM等流程。
二、软件的安装
1. Conda软件安装
conda是常用的软件安装和管理软件,操作简单、便捷。
https://mirrors.tuna.tsinghua.edu.cn/
conda软件的下载,可下载miniconda或Anaconda。
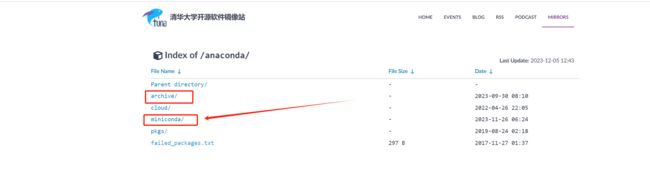
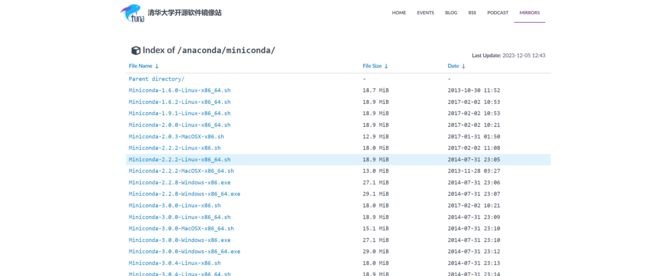
2. miniconda(下载对应的版本)
3. Anaconda(下载对应的版本)

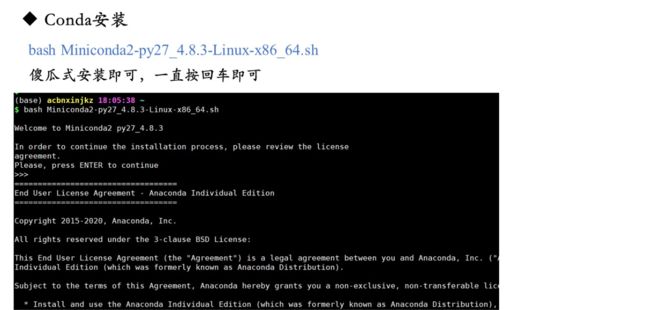
4. 软件的安装
5. 添加常用镜像
若是不能使用,可以自己百度一下进行搜索即可。
## Conda常使用的镜像
# 下面这四行配置清华大学的bioconda的channel地址,国内用户推荐
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
conda config --set show_channel_urls yes
# 中科大镜像源
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/cloud/msys2/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/cloud/bioconda/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/cloud/menpo/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/cloud/
# 阿里镜像源
conda config --add channels https://mirrors.aliyun.com/pypi/simple/
# 豆瓣镜像
conda config --add channels http://pypi.douban.com/simple/
#中国科学技术大学 USTC Mirror
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.ustc.edu.cn/anaconda/cloud/conda-forge/
6. 创建生信环境
若是你担心自己base环境被破坏,那么就安装自己对于的小环境即可。
## 创建环境
conda create -n env_name python=x.x
## 删除环境
conda remove -n env_name -all
## 激活
conda activate env_name
##
source activate env_name
## 关闭
conda deactivate
查看环境中的软件
# 查看指定环境下安装的package
## 查看指定环境下安装的package
conda list -n env_name
## 安装指定环境下某个package
conda install -n env_name [package]
## 删除指定环境下某个package
conda remove -n env_name [package]
## 更新指定环境下某个package
conda update -n env_name [package]
三、生信比对软件的安装
- 安装mamba软件,mamba相对于conda安装软件,速度更快,也更容易安装。
conda install -y mamba
比对所需的软件…
- hista2
- Stringtie
- subread
- samtools
- fastp
mamba install hisat2
mamba install stringtie
mamba install samtools
mamba install subread
mamba install fastp
- 使用源码安装
直接下载对应的软件源码,解压后进行安装。

四、数据的下载
公共数据库的下载,可直接在NCBI中下载,或是使用自己测的数据即可。若你想使用公共数据库的数据,可以我们前面的教程转录组数据的下载。
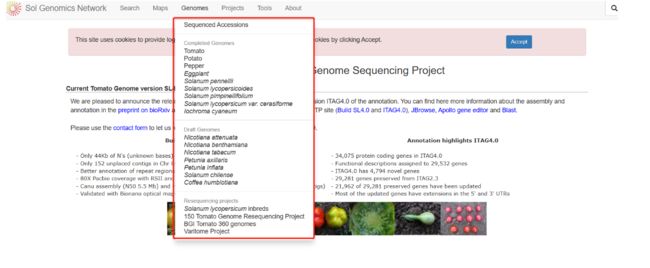
五、基因组的下载
- 大部分的作物有自己基因组注释网址,我们需要自己的去寻找
模式植物中,拟南芥、番茄、 烟草等都有自己的基因组网址。
茄科类作物基因组:https://solgenomics.net/organism/solanum_lycopersicum/genome
– NCBI 中下载基因组文件
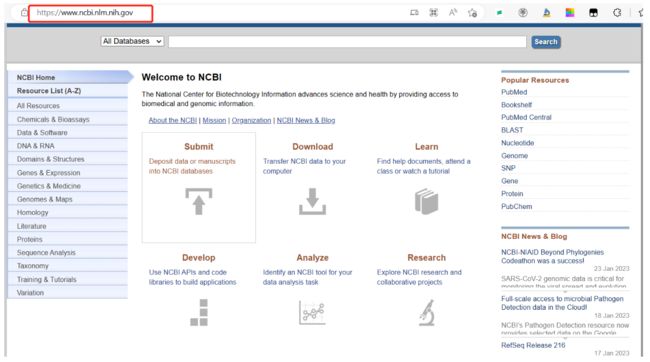
- 如果自己的物种基因组没有单独的网址,如何做呢?
可以根据NCBI中进行下载
步骤:
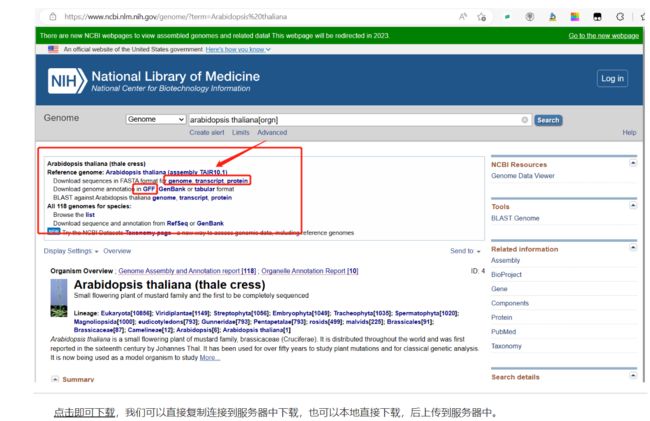
- 进入NCBI官网(https://www.ncbi.nlm.nih.gov/)
- 输入需要寻找的基因组名称 (可以是作物名或是拉丁名)
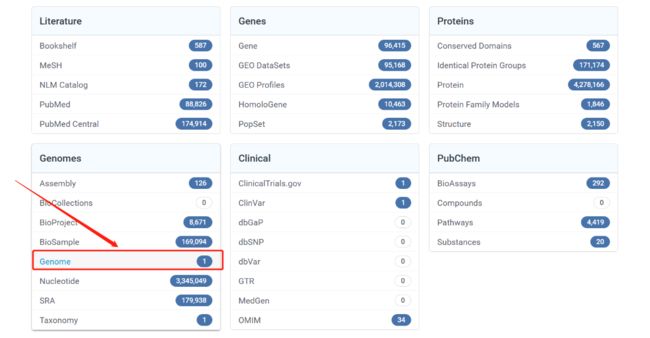
在此界面就是我们的作物的基因组信息,有版本信息,geneome,transcript,protein,GFF,GenBank等信息。
六、数据过滤和质控检测
-
使用
FastP,主要是简单、便捷。
软件官网:[https://github.com/OpenGene/fastp](https://github.com/OpenGene/fastp}
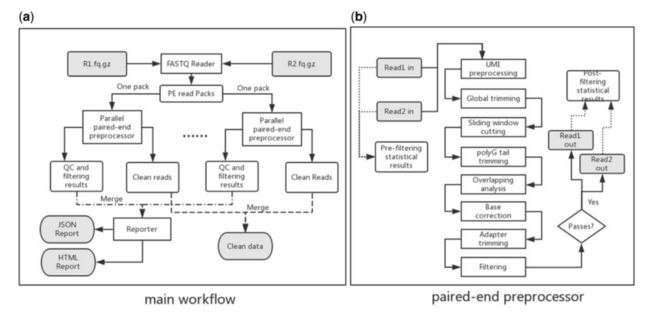
-
FastQC进行质量评估
FastQC旨在提供一种简单的方法,对来自高通量测序管道的原始序列数据做一些质量控制检查。它提供了一套模块化的分析,你可以用它来快速了解你的数据是否有任何问题,在做任何进一步的分析之前,你应该注意到这些问题。
在处理任何样品之前的第一步是分析数据的质量。在fastq文件内有质量信息,指的是每个碱基调用的准确性(置信度%)。FastQC查看样品序列的不同方面,以确定任何影响结果的不规则或特征(适配器污染、序列重复水平等)。
本教程详细教程:一个转录组上游分析流程 | Hisat2-Stringtie****
到这里,本期教程到这里就结束了。很多的参数需要结合自己的数据进行调整。
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
-
WGCNA分析 | 全流程分析代码 | 代码四
-
WGCNA分析 | 全流程分析代码 | 代码五(最新版本)
4. 精美图形绘制教程
- 精美图形绘制教程
5. 转录组分析教程
转录组上游分析教程[零基础]
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!