晶格动力学程序 GULP
大家好,我是木南
分子动力学软件诸多,包括:
1. LAMMPS
LAMMPS即Large-scale Atomic/Molecular Massively Parallel Simulator,可以翻译为大规模原子分子并行模拟器,主要用于分子动力学相关的一些计算和模拟工作,一般来讲,分子动力学所涉及到的领域,LAMMPS代码也都涉及到了。LAMMPS可以支持包括气态,液态或者固态相形态下、各种系综下、百万级的原子分子体系,并提供支持多种势函数。
2. DL_POLY
DL_POLY是串行和并行分子动力学模拟软件包。DL_POLY目前有两个版本。DL_POLY_2是原始版本,用复制数据的方法并行化,适用于在100个处理器上模拟三万个原子的情况;DL_POLY_3的并行化使用区域分解,适用于在8至1024个处理器上,模拟百万量级的原子。对于一个DL_POLY许可,同时提供两个版本。
3. SIESTA
SIESTA是一个可以免费索取许可的学术计算软件,用于分子和固体的电子结构计算和分子动力学模拟。SIESTA 使用标准的Kohn-Sham 自恰密度泛函方法,计算使用完全非局域形式(Kleinman-Bylander)的标准守恒赝势。基组是数值原子轨道的线性组合(LCAO)。它允许任意个角动量,多个zeta,极化和截断轨道。计算中把电子波函和密度投影到实空间网格中,以计算Hartree和XC势,及其矩阵元素。
4. PWscf
PWscf计算软件是意大利理论物理研究中心发布的Quantum-ESPRESSO计算软件包中的两大模块之一。Quantum-ESPRESSO软件包的开发遵守GNU自由软件的协议,是基于密度泛函理论,应用平面波基组和赝势方法的第一性原理计算软件。先前由于计算软件的落后而使得一些有用的方法如线性响应、超软赝势CP分子动力学(MD)方法等,受到了应用上的阻碍,这个软件的发布正是基于这种情况,从而提供了应用这些方法的一个平台。它包括两大模块:PWscf和CPMD。
5. ABINIT
ABINIT的主程序使用赝势和平面波,用密度泛函理论计算总能量,电荷密度,分子和周期性固体的电子结构,进行几何优化和分子动力学模拟,用TDDFT(对分子)或GW近似(多体微扰理论)计算激发态。此外还提供了大量的工具程序。程序的基组库包括了元素周期表1-109号所有元素。ABINIT适于固体物理,材料科学,化学和材料工程的研究,包括固体,分子,材料的表面,以及界面,如导体、半导体、绝缘体和金属。
见于网上关于GULP资料不多,所以先从GULP介绍:
GULP是由澳大利亚科廷大学J.D. Gale为首的研究团队开发的,最初设计的目的是拟合力场,现已成为模拟凝聚态物质的通用程序。
GULP软件现已发展到5.2版本,其使用Fortran编译器,可运行在Linux/Unix系统下,但不提供任何Windows版本的技术支持。
下载网址:GULP - Home
案例网址:GULP - Examples
帮助手册网址:http://gulp.curtin.edu.au/gulp/help/help_52_txt.html
安装方法:在Linux系统中输入“./mkgulp”即可!
可对材料模拟的尺度类型:
- 零维材料(分子、团簇和缺陷)
- 一维材料(聚合物和位错)
- 二维材料(板、表面、晶界和界面)
- 三维材料(块体材料)
1. 零维材料:一般是指纳米材料。即是指在三维空间中至少有一维处于纳米尺寸的材料,如富勒烯C60。

2. 一维材料:是指线状材料。如碳纳米管,在一个方向上原子长程规则排列到厘米量级,在另外两个方向上只有很少原子排列的材料。

3. 二维材料:是指带状材料(片层材料)。在空间两个尺度上远远大于纳米量级,一个尺度上仍为纳米量级。如单片层状石墨。

4. 三维材料:就是日常生活中用的材料,如金刚石。

部分内容转自网络:https://www.zhihu.com/question/46430290/answer/102273965
主要功能:
- 能量最小化
- 晶体特性(弹性常数、体积模量、剪切模量、杨氏模量、泊松比、压缩比、介电常数等)
- 热力学性质(热容、熵、焓、自由能、声子频率、声子态密度等)
- 缺陷计算(空位和间隙缺陷、表面能和吸附能等)
- 力场拟合(弛豫拟合、经验拟合、从头算能量拟合等)
- 结构分析(键长、键角、扭转角和距离等)
- 过渡状态
- 分子动力学模拟(壳层模型和呼吸壳层模型,系综:NVT、NVE和NPT等)
- 蒙特卡罗模拟(物质的旋转或位移)
- 其他功能
此外,GULP还可为其他一些常用的模拟软件生成输入文件,例如:MARVIN、Materials Studio、G-Vis和SIESTA等,其中Materials Studio软件也含有GULP模块。
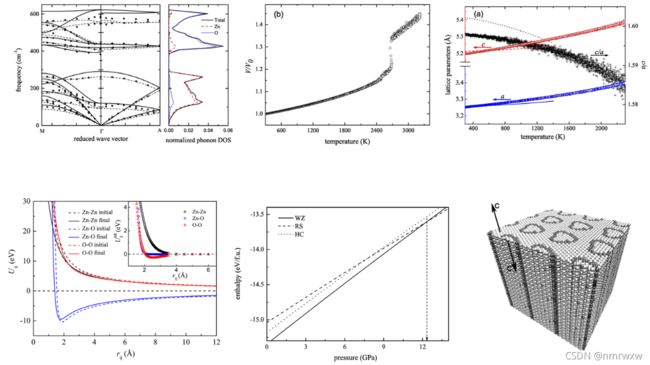
上述图片来自于文献:
New Ab Initio Based Pair Potential for Accurate Simulation of Phase Transitions in ZnO
链接:https://doi.org/10.1021/jp411308z



GULP的主要优势:
- 相对于传统算法,GULP采用了对称适应算法
- 输入文件简化,变量个数少,也使等效的相互作用的计算不必重复
- 计算能力有数量级的改变,提高了计算效率,大大减少了计算时间
- 主要由晶体结构和力场两个部分组成,输入相应的参数或数据即可运行
可用的原子间势函数形式:
- Buckingham
- Lennard-Jones
- Morse
- Spring (core-shell)
- Breathing shell harmonic
- Stillinger-Weber
- Four-body torsional potential
- ReaxFF

以上是我们分享的一些经验,或有不足,欢迎大家指出并留言!最后,感谢功能新材料高压物性研究小组以及本课题组所有人的支持!
欢迎大家交流!扫码即可关注微信公众号:原子与分子模拟
