【算法详解】STitch3D:空间转录组数据和单细胞RNA测序数据共同建模的空转数据对齐和整合算法,实现更高分辨率的3D结构重建
目录
0 参考文献
1 模型特色
2 算法解析
2.1 模型概述
2.2 获得隐空间表示
2.3 获得细胞类型比例
2.4 处理批次效应
0 参考文献
STitch3D原论文:Construction of a 3D whole organism spatial atlas by joint modelling of multiple slices with deep neural networks
1 模型特色
生物体器官具有复杂的三维结构,生理过程的进行也很少在二维平面发生,然而现存的多数工具和方法只能提供对于生物组织二维切片的单独分析,这限制了对细胞的三维排列、细胞间相互作用的理解。为了改善这种现象,许多用于空间组学数据对齐或数据整合的方法应运而生。
此前也有为了解决类似问题而整合多切片空转数据的方法例如STAligner、SPIRAL等。而在这篇博客中将介绍一种更新的方法STitch3D。
STitch3D与之前方法的一种区别在于,STitch3D同时使用空转数据和单细胞RNA测序数据进行建模,而之前的方法大多只使用空间转录组数据。
由于受到技术限制,目前空间转录组数据分辨率无法达到单细胞级别,在之前的博客中也有提到,空间转录组测量出的基因表达矩阵每一行代表一个spot,而一个spot将包含多个细胞。STitch3D方法通过引入单细胞RNA测序数据,实现了对于spot的细胞类型分解,从而使基于STicth3D重建的3D结构具有“更高的分辨率”,可以同时完成识别空间区域(区分不同区域)和推断细胞类型分布两种任务。后者在上面提到的方法中是不能实现的。
2 算法解析
2.1 模型概述
STitch3D是一个基于图注意力深度学习网络、使用多个切片的空转数据重建3D组织结构的方法。模型结构如下图所示。
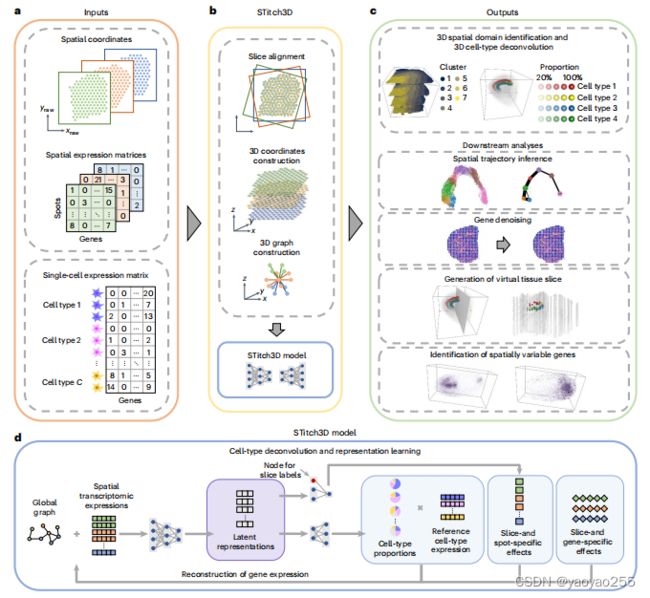
模型的输入是多个空转切片和一个对应的单细胞RNA测序(scRNA-seq)参考。
预处理步骤需要先将这些空间转录组切片进行对齐,获得对齐后的新的2D坐标,从而根据这些新坐标构建3D坐标,再使用3D坐标构建3D邻接图。与同样需要构建邻接图的STAligner方法不同点在于,STAligner是在单个切片内构建2D邻接图,由于STAligner并没有提前进行切片对齐,因此基于原始坐标构建3D邻接图是不合理的。
在此之后,这些数据使用STitch3D模型进行整合,在共享隐空间中获得每个spot的低维表示,并用于空间域识别和细胞类型分解。这个隐空间可以分离有意义的生物学区别并减小批次效应的影响。
最终可以得到每个spot的低维表示和细胞类型占比。
2.2 获得隐空间表示
假设空转切片有S个,用![]() 表示切片索引。
表示切片索引。
第s个切片的spot数量为 ,基因表达矩阵为
,基因表达矩阵为![]() ,其中
,其中![]() 为spot的索引,
为spot的索引,![]() 为基因索引。
为基因索引。
在对应的scRNA-seq参考中,不同类型细胞的基因表达情况组成的基因表达矩阵为![]() ,其中
,其中![]() 为细胞类型索引,矩阵中每一行代表一种类型的细胞的记忆表达情况,有约束
为细胞类型索引,矩阵中每一行代表一种类型的细胞的记忆表达情况,有约束![]() 。
。
STitch3D首先使用ICP或PASTE算法对多个切片进行对齐,这里也可以替换为其他可以实现对齐的算法。这之后给每一个spot一个z轴坐标,z轴坐标描绘了切片之间的距离,可以根据切片真实距离设置。
根据每个spot的3D坐标,构建3D邻接图。默认情况下,将相互之间距离小于同切片两spot最小间距1.1倍距离的两个spot认为是邻接的。
在输入模型之前,先将所有S张切片顺序拼接起来,以![]() 作为拼接后的基因表达矩阵,
作为拼接后的基因表达矩阵,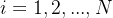 作为新的spot索引,其中
作为新的spot索引,其中![]() 为S张切片的spot总数。以
为S张切片的spot总数。以![]() 表示之前计算的3D邻接图(矩阵),若索引为i和j的spot是邻接的则对应位置为1,否则为0。
表示之前计算的3D邻接图(矩阵),若索引为i和j的spot是邻接的则对应位置为1,否则为0。
随后将数据输入模型,通过一个基于图注意力的深度学习网络![]() 得到隐空间表示
得到隐空间表示![]() ,其中p为隐空间维度。
,其中p为隐空间维度。
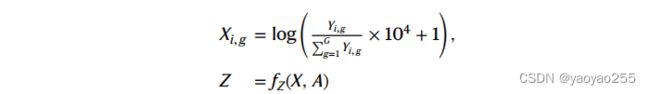
2.3 获得细胞类型比例
得到了spot的隐空间表示,就可以根据隐空间表示进一步得到spot的细胞类型比例![]() ,其中
,其中![]() 表示细胞类型c在第i个spot中的占比,且有
表示细胞类型c在第i个spot中的占比,且有![]() 。
。![]() 为第i个spot的低维表示。
为第i个spot的低维表示。
这里假设细胞类型比例可以与该spot的低维表示有关,因此使用神经网络![]() ,通过
,通过![]() 得到该spot的细胞类型比例。
得到该spot的细胞类型比例。
由于计算隐空间表示Z使用到了空间关系A,因此计算出的细胞类型比例也包含空间关系信息。
2.4 处理批次效应
由于测量空转数据和scRNA-seq的技术不同,因此两种数据之间存在着不可避免的扰动,并且在不同切片之间也会存在批次效应。
为了处理批次效应,STitch3D引入![]() 和
和![]() 两个(组)量,其中
两个(组)量,其中![]() 表示切片-spot特异的影响,
表示切片-spot特异的影响,![]() 表示切片-基因特异的影响。
表示切片-基因特异的影响。
结合新定义的量,STitch3D以下面方式,通过细胞类型比例和特定类型细胞的基因表达情况重建每个spot的基因表达向量。

其中 是第i个spot中观察到的基因表达总数(原始Y矩阵第i行元素之和)。
是第i个spot中观察到的基因表达总数(原始Y矩阵第i行元素之和)。
假设![]() 与基因表达数据和spot所在切片有关,因此
与基因表达数据和spot所在切片有关,因此![]() 通过一个以第i个spot的低维表示和所在切片的标签作为输入的网络的输出得到。如下所示
通过一个以第i个spot的低维表示和所在切片的标签作为输入的网络的输出得到。如下所示
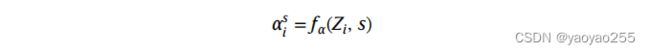
![]() 则作为可训练参数。
则作为可训练参数。
这部分的损失函数如下
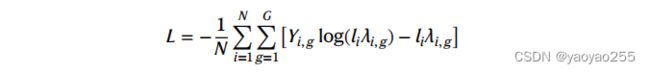
STitch3D使用一个正则化器来保留切片间想要的生物学区别,正则化器如下

其中基于图注意力网络的切片特异的网络![]() 用于通过spot的低维表示重建
用于通过spot的低维表示重建 。
。
整体的损失函数定义为![]() ,其中
,其中![]() 为超参数。
为超参数。