GOplot包介绍
GOplot包用于生物数据的可视化。更确切地说,该包将表达数据与功能分析的结果整合并进行可视化。但是要注意该包不能用于执行这些分析,只能把分析结果进行可视化。在所有科学领域,由于空间限制和结果所需的简洁性,切实地去描述事物很难,所以需要将信息进行可视化,使用图片来传达信息。精心设计的图形能在更小的空间提供更多的信息。该包的设想就是能让用户快速检查大量数据,揭示数据的趋势和找出数据中的模式和相关性。
论文图表基本规范
学术图表的基本配色方法
数据可视化基本套路总结
数据可视化可以帮助我们去寻找生物问题的答案,也可以对某一假设进行判断,甚至可能发现不同的角度来调查不同的问题。并且该包的画图函数是以数据的层次结构为基础进行开发的,从整体数据开始,以所选基因和对应通路的子集结束。
让我们用例子来具体解释。
举例说明
我们调用GOplot自带的数据,该数据来自于GEO的GSE47067,包含来自两个组织(脑和心脏)的内皮细胞的转录组信息,更多信息详见Nolan等人的论文https://www.ncbi.nlm.nih.gov/pubmed/23871589,然后将数据进行标准化处理并寻找差异表达的基因,再使用DAVID功能注释工具(DAVID注释数据更新比较慢,现在已经不太推荐,建议用去东方,最好用的在线GO富集分析工具和这个只需一步就可做富集分析的网站还未发表就被CNS等引用超过350次进行富集分析, 一文掌握GSEA,超详细教程)对差异表达基因进行基因注释(adjusted p-value < 0.05)和功能富集分析。该数据集包含以下五类数据:
| 名称 | 描述 | 数据集大小 |
|---|---|---|
| EC$eset | 进行标准化后的脑和心脏内皮细胞(3次重复)基因表达量 | 20644 x 7 |
| EC$genelist | 差异表达基因(adjusted p-value < 0.05) | 2039 x 7 |
| EC$david | 用DAVID对差异基因进行功能富集分析的结果 | 174 x 5 |
| EC$gene | 基因和logFC | 37 x 2 |
| EC$process | 选择的富集生物过程的特征向量 | 7 |
了解数据格式
我们希望查看差异表达基因的GO富集通路,但在我们开始画图之前我们需要提供符合格式需求的数据。通常来说,画图所需的数据是自己提供,但该包内有一个函数circle_dat可以帮我们处理数据格式。circle_dat能合并所选择的基因的功能富集分析结果及其logFC值,主要是用于差异表达基因。circle_dat的使用很简单,只要读入两个数据即可。第一个数据包含功能富集分析结果,至少有四列(功能富集分析类别、通路、基因、adjusted p-value)。第二个数据是所选基因的及其logFC,该数据可以是来源limma统计分析的结果 (生信宝典注:一定注意两个文件基因的命名方式要一致,比如都是Gene symbol)。我们用实例来看以上提到的数据格式。
#安装已发布的稳定版本
#install.packages('GOplot')
#安装github上的开发版本
#install_github('wencke/wencke.github.io')
#载入包
library(GOplot)
#读入包内自带的数据
data(EC)
#查看功能富集分析结果的数据格式
head(EC$david)
## Category ID Term
## 1 BP GO:0007507 heart development
## 2 BP GO:0001944 vasculature development
## 3 BP GO:0001568 blood vessel development
## 4 BP GO:0048729 tissue morphogenesis
## 5 BP GO:0048514 blood vessel morphogenesis
## 6 BP GO:0051336 regulation of hydrolase activity
## Genes
## 1 DLC1, NRP2, NRP1, EDN1, PDLIM3, GJA1, TTN, GJA5, ZIC3, TGFB2, CERKL, GATA6, COL4A3BP, GAB1, SEMA3C, MKL2, SLC22A5, MB, PTPRJ, RXRA, VANGL2, MYH6, TNNT2, HHEX, MURC, MIB1, FOXC2, FOXC1, ADAM19, MYL2, TCAP, EGLN1, SOX9, ITGB1, CHD7, HEXIM1, PKD2, NFATC4, PCSK5, ACTC1, TGFBR2, NF1, HSPG2, SMAD3, TBX1, TNNI3, CSRP3, FOXP1, KCNJ8, PLN, TSC2, ATP6V0A1, TGFBR3, HDAC9
## 2 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, EFNB2, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
## 3 GNA13, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, FOXO1, GJA5, TGFB2, WARS, CERKL, APOE, CXCR4, ANG, SEMA3C, NOS2, MKL2, FGF2, RAPGEF1, PTPRJ, RECK, VASH1, PNPLA6, THY1, MIB1, NUS1, FOXC2, FOXC1, CAV1, CDH2, MEIS1, WT1, CDH5, PTK2, FBXW8, CHD7, PLCD1, PLXND1, FIGF, PPAP2B, MAP2K1, TBX4, TGFBR2, NF1, TBX1, TNNI3, LAMA4, MEOX2, ECSCR, HBEGF, AMOT, TGFBR3, HDAC7
## 4 DLC1, ENAH, NRP1, PGF, ZIC2, TGFB2, CD44, ILK, SEMA3C, RET, AR, RXRA, VANGL2, LEF1, TNNT2, HHEX, MIB1, NCOA3, FOXC2, FOXC1, TGFB1I1, WNT5A, COBL, BBS4, FGFR3, TNC, BMPR2, CTNND1, EGLN1, NR3C1, SOX9, TCF7L1, IGF1R, FOXQ1, MACF1, HOXA5, BCL2, PLXND1, CAR2, ACTC1, TBX4, SMAD3, FZD3, SHANK3, FZD6, HOXB4, FREM2, TSC2, ZIC5, TGFBR3, APAF1
## 5 GNA13, CAV1, ACVRL1, NRP1, PGF, IL18, LEPR, EDN1, GJA1, CDH2, MEIS1, WT1, TGFB2, WARS, PTK2, CERKL, APOE, CXCR4, ANG, SEMA3C, PLCD1, NOS2, MKL2, PLXND1, FIGF, FGF2, PTPRJ, TGFBR2, TBX4, NF1, TBX1, TNNI3, PNPLA6, VASH1, THY1, NUS1, MEOX2, ECSCR, AMOT, HBEGF, FOXC2, FOXC1, HDAC7
## 6 CAV1, XIAP, AGFG1, ADORA2A, TNNC1, TBC1D9, LEPR, ABHD5, EDN1, ASAP2, ASAP3, SMAP1, TBC1D12, ANG, TBC1D14, MTCH1, TBC1D13, TBC1D4, TBC1D30, DHCR24, HIP1, VAV3, NOS1, NF1, MYH6, RICTOR, TBC1D22A, THY1, PLCE1, RNF7, NDEL1, CHML, IFT57, ACAP2, TSC2, ERN1, APAF1, ARAP3, ARAP2, ARAP1, HTR2A, F2R
## adj_pval
## 1 0.000002170
## 2 0.000010400
## 3 0.000007620
## 4 0.000119000
## 5 0.000720000
## 6 0.001171166
#查看基因的数据格式
head(EC$genelist)
## ID logFC AveExpr t P.Value adj.P.Val B
## 1 Slco1a4 6.645388 1.2168670 88.65515 1.32e-18 2.73e-14 29.02715
## 2 Slc19a3 6.281525 1.1600468 69.95094 2.41e-17 2.49e-13 27.62917
## 3 Ddc 4.483338 0.8365231 65.57836 5.31e-17 3.65e-13 27.18476
## 4 Slco1c1 6.469384 1.3558865 59.87613 1.62e-16 8.34e-13 26.51242
## 5 Sema3c 5.515630 2.3252117 58.53141 2.14e-16 8.81e-13 26.33626
## 6 Slc38a3 4.761755 0.9218670 54.11559 5.58e-16 1.76e-12 25.70308
了解了两个输入数据格式后,就可以用cirlce_dat函数来生成画图数据了。
# 生成画图所需的数据格式
circ <- circle_dat(EC$david, EC$genelist)
head(circ)
## category ID term count genes logFC adj_pval
## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
## zscore
## 1 -0.8164966
## 2 -0.8164966
## 3 -0.8164966
## 4 -0.8164966
## 5 -0.8164966
## 6 -0.8164966
circ对象有八列数据,分别是
category:BP(生物过程),CC(细胞组分)或 MF(分子功能)
ID: GO id(可选列, 想使用不基于GO id的功能分析工具,可以不选ID列;这里的ID也可以是KEGG ID)
term:GO通路
count:每个通路的基因个数
gene:基因名 - logFC:每个基因的logFC值
adj_pval:adjusted p值,adj_pval<0.05的通路被认为是显着富集的
zscore:zscore不是指统计学的标准化方式,而是一个很容易计算的值,来估计生物过程(/分子功能/细胞成分)更可能降低(负值)或增加(正值)。计算方法就是上调基因的数量减去下调基因的数量再除以每个通路基因数目的平方根
画图
GOBar–条形图
最开始查看数据时,我们希望能从图中展示尽可能多的通路,并且也希望能找到有价值的通路,因此需要一些参数来评估重要性。条形图经常用于描述样本数据,故而我们可以用GOBar函数能快速创建一个好看的的条形图。
首先直接生成一个简单的条形图,横轴是GO Terms,根据它们的zscore对条进行排序;纵轴是 -log(adj p-value);颜色表示的是zscore,蓝色表示z-score是负值,在对应通路基因表达更可能下降,红色表示z-score是正值,在对应通路基因表达更可能升高。如果需要,可以通过将参数order.by.zscore设置为FALSE来更改顺序,在这种情况下条形图基于它们的显著性进行排序。
# 生成简单的条形图
GOBar(subset(circ, category == 'BP'))

#GOBar(subset(circ, category == 'BP',order.by.zscore=FALSE))
另外,通过更改display参数来根据通路的类别来绘制条形图。
#根据通路的类别来绘制条形图
GOBar(circ, display = 'multiple')

添加标题,并使用参数zsc.col更改zscore的颜色。
# Facet the barplot, add a title and change the colour scale for the z-score
GOBar(circ, display = 'multiple', title = 'Z-score coloured barplot', zsc.col = c('yellow', 'black', 'cyan'))

条形图是很常见的,也很容易理解,但我们可以使用气泡图来显示数据更多信息。
GOBubble–泡泡图
横轴是zscore;纵轴是-log(adj p-value),类似于条形图,越高表示富集越显着;圆的面积与对应通路的的基因数(circ$count)成正比;颜色对应于该通路所对应的类别,绿色生物过程,红色是细胞组分,蓝色是分子功能。可通过输入?GOBubble查看GOBubble函数的帮助页面来更改图片的所有参数。在默认情况下,每个圆标有对应的GO ID,右侧也会随之显示GO ID和GO term对应关系的表。可通过设置参数table.legend为FALSE来隐藏它。如果要显示通路描述,请设置参数ID为FALSE。不过由于空间有限和圆重叠,并非所有圆都被标记,只显示了-log(adj p-value) > 3(默认是5)的通路。
# 生成泡泡图,并展示-log(adj p-value) > 3 的通路的
GO IDGOBubble(circ, labels = 3)

若给泡泡图要添加标题,或指定圆圈的颜色并单独展示各类别的通路并更改展示的GO ID阈值,可添加以下参数:
GOBubble(circ, title = 'Bubble plot', colour = c('orange',
'darkred', 'gold'), display = 'multiple', labels = 3)
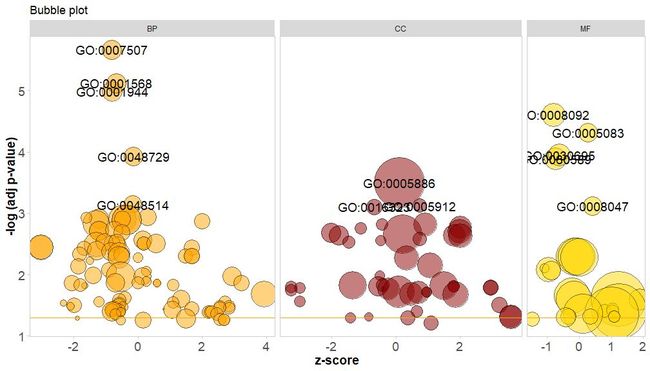
通过将参数bg.col设置为TRUE,为通路的类别的背景着色。
GOBubble(circ, title = 'Bubble plot with background colour', display = 'multiple', bg.col = T, labels = 3)

新版本的包中包含一个新函数reduce_overlap,该函数可以减少冗余项的数量,即能删除基因重叠大于或等于设定阈值的所有通路,只将每组的一个通路作为代表保留,而不考虑GO所有通路的展示。通过减少冗余项的数量,图的可读性(如气泡图)显着改善。
# reduce_overlap,参数设置为0.75
reduced_circ <- reduce_overlap(circ, overlap = 0.75)
GOBubble(reduced_circ, labels = 2.8)

GOCircle–圈图展示基因功能富集分析结果
虽然展示所有信息的图有助于我们发现哪些通路最有意义,不过实际情况还是取决于你想要用数据确认的假设和想法,最重要的通路也不一定是你感兴趣的。因此,在手动选择一组有价值的通路(EC$process)后,我们需要一张图为我们展示此组特定通路的更详细的信息。不过通过呈现这些图能得出一个问题:有时很难解释zscore提供的信息。毕竟这个计算方法并不通用,如上所示,它仅仅是上调基因的数量减去下调基因的数量除以每个通路基因数目的平方根,用GOCircle得出的图也强调了这一事实。
圈图外圈的圆用散点展示了每个通路的基因的logFC值。红色圆圈表示上调和蓝色表示下调。可以使用参数lfc.col更改颜色。这也解释了为什么在某些情况下,非常重要的通路具有接近零的zscore。zscore为零并不意味着该通路不重要。它只是表明zscore是粗略的衡量标准,因为显然zscore也没有考虑生物过程中单个基因的功能水平和激活依赖性。
GOCircle(circ)
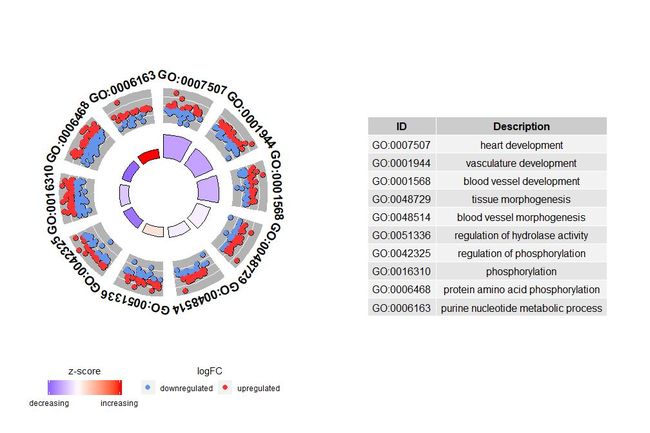
nsub参数可是设置数字或字符向量。如果它是字符向量,则它包含要显示的GO ID或通路;
# 生成特定通路的圈图
IDs <- c('GO:0007507', 'GO:0001568', 'GO:0001944', 'GO:0048729', 'GO:0048514', 'GO:0005886', 'GO:0008092', 'GO:0008047')
GOCircle(circ, nsub = IDs)

如果nsub是数字向量,则该数字定义显示的个数。它从输入数据帧的第一行开始。这种可视化仅适用于较小的数据。最大通路数默认为12。虽然通路数量减少,但显示的信息量会增加。
# 圈图展示数据前十个通路
GOCircle(circ, nsub = 10)

GOChord–圈图展示基因和通路之间的关系
GOChord能展示了所选基因和通路之间的关系和基因的logFC。首先需要输入一个矩阵,可以自己构建0-1矩阵,也可以使用函数chord_dat构建。该函数有三个参数:data,genes和process,其中最后两个参数至少要有一个参数。然后函数circle_dat将表达数据与功能分析的结果相结合。
条形图和气泡图可以让您对数据有第一印象,现在,可以选择了一些我们认为有价值的基因和通路,尽管GOCircle添加了一个层来显示基因在通路的表达值,但它缺乏单个基因和多个通路之间关系的信息。要弄清楚某些基因是否与多个过程相关联并不容易。GOChord就弥补了GOCircle的缺陷。生成的数据行是基因,列是通路,“0”表示该基因未被分配到该通路,“1”正相反。
# 找到感兴趣的的基因,这里我们以EC$genes为例
head(EC$genes)
## ID logFC
## 1 PTK2 -0.6527904
## 2 GNA13 0.3711599
## 3 LEPR 2.6539788
## 4 APOE 0.8698346
## 5 CXCR4 -2.5647537
## 6 RECK 3.6926860
# 获得感兴趣基因的通路
EC$process
## [1] "heart development" "phosphorylation"
## [3] "vasculature development" "blood vessel development"
## [5] "tissue morphogenesis" "cell adhesion"
## [7] "plasma membrane"
# 使用chord_dat构建矩阵
chord <- chord_dat(circ, EC$genes, EC$process)
head(chord)
## heart development phosphorylation vasculature development
## PTK2 0 1 1
## GNA13 0 0 1
## LEPR 0 0 1
## APOE 0 0 1
## CXCR4 0 0 1
## RECK 0 0 1
## blood vessel development tissue morphogenesis cell adhesion
## PTK2 1 0 0
## GNA13 1 0 0
## LEPR 1 0 0
## APOE 1 0 0
## CXCR4 1 0 0
## RECK 1 0 0
## plasma membrane logFC
## PTK2 1 -0.6527904
## GNA13 1 0.3711599
## LEPR 1 2.6539788
## APOE 1 0.8698346
## CXCR4 1 -2.5647537
## RECK 1 3.6926860
示例中我们传递了两个参数,若只指定genes参数,则结果是所选基因列表和具有至少一个指定基因的所有过程构建0-1矩阵;若只指定了process参数,则结果是所有基因生成0-1矩阵,这些基因分配给列表中的至少一个过程。要注意只指定genes和process参数可能会导致0-1矩阵很大,从而导致可视化结果混乱。

head(circ)
## category ID term count genes logFC adj_pval
## 1 BP GO:0007507 heart development 54 DLC1 -0.9707875 2.17e-06
## 2 BP GO:0007507 heart development 54 NRP2 -1.5153173 2.17e-06
## 3 BP GO:0007507 heart development 54 NRP1 -1.1412315 2.17e-06
## 4 BP GO:0007507 heart development 54 EDN1 1.3813006 2.17e-06
## 5 BP GO:0007507 heart development 54 PDLIM3 -0.8876939 2.17e-06
## 6 BP GO:0007507 heart development 54 GJA1 -0.8179480 2.17e-06
## zscore
## 1 -0.8164966
## 2 -0.8164966
## 3 -0.8164966
## 4 -0.8164966
## 5 -0.8164966
## 6 -0.8164966
# Generate the matrix with a list of selected genes
chord_genes <- chord_dat(data = circ, genes = EC$genes)
head(chord_genes)
## heart development vasculature development blood vessel development
## PTK2 0 1 1
## GNA13 0 1 1
## LEPR 0 1 1
## APOE 0 1 1
## CXCR4 0 1 1
该图表是为了展示较小的高维数据的子集。主要可以调整两个参数:gene.order和nlfc。genes参数可指定为’logFC’,‘alphabetical’,‘none’。实际上,我们一般指定genes参数为logFC;nlfc参数是这个函数最重要的参数之一,因为它能处理每个基因有0个或多个logFC值怎么在矩阵呈现。故而我们应该指定参数来避免错误。
例如,如果有一个没logFC值的矩阵,则必须设置nlfc=0;或者在多个条件或批次对基因进行差异表达分析,这时每个基因包含多个logFC值,需要设置nlfc=logFC列数。默认值为“1”,因为认为大多数时候每个基因只有一个logFC值。用space参数定义表示logFC的彩色矩形之间的空间。gene.size参数规定基因名字字体大小,gene.space规定基因名字间的空间大小。
chord <- chord_dat(data = circ, genes = EC$genes, process = EC$process)
GOChord(chord, space = 0.02, gene.order = 'logFC', gene.space = 0.25, gene.size = 5)
## Warning: Using size for a discrete variable is not advised.
## Warning: Removed 7 rows containing missing values (geom_point).

可根据logFC值设定gene.order=‘logFC’,对基因按照logFC值进行排序。有时图片会变得有点拥挤,可以通过使用limit参数自动执行减少显示的基因或通路的数量。Limit是具有两个截止值的向量(默认值是c(0,0))。第一个值规定了基因必须分配的最少通路个数。第二个值确定分配给通路的基因个数。
# 仅显示分配给至少三个通路的基因
GOChord(chord, limit = c(3, 0), gene.order = 'logFC')
## Warning: Using size for a discrete variable is not advised.
## Warning: Removed 7 rows containing missing values (geom_point).

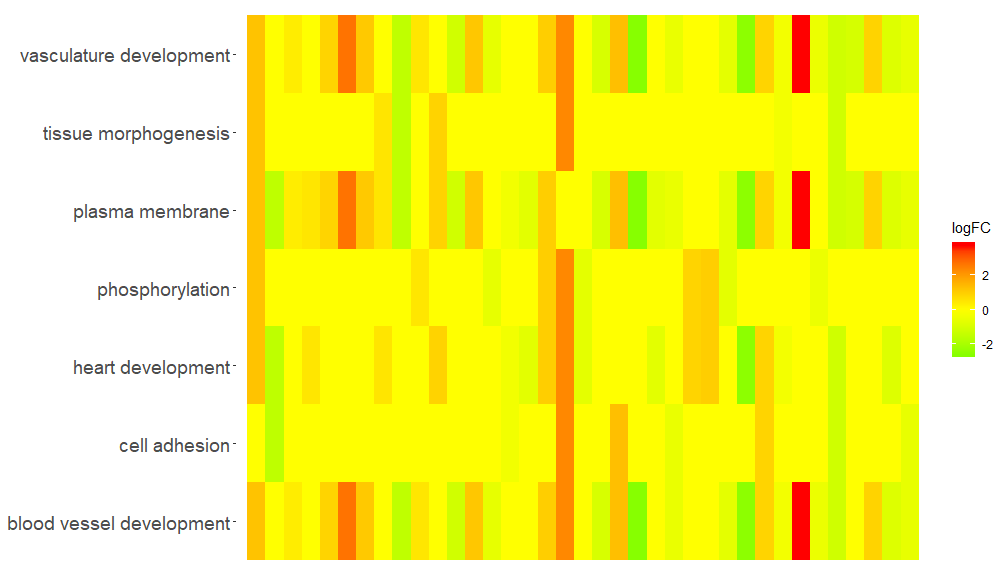
GOHeat–热图展示基因和通路
GOHeat函数能用热图展示基因和通路之间的关系,类似于GOChord。横向展示生物过程,纵向展示基因。每列被分成小的矩形,颜色一般取决于logFC值。另外具有富集到相似功能通路的基因被聚类。热图颜色选择有两种模式,具体取决于nlfc参数。如果nlfc = 0,则颜色为每个基因所富集到的通路个数。详见例子:
# First, we use the chord object without logFC column to create the heatmap
GOHeat(chord[,-8], nlfc = 0)

GOHeat(chord[,-8])
在nlfc = 1的情况下,颜色对应于基因的logFC
GOHeat(chord, nlfc = 1, fill.col = c('red', 'yellow', 'green'))
GOCluster–Golden eye
GOCluster功能背后的想法是尽可能多地显示信息。这是一个例子:
GOCluster(circ, EC$process, clust.by = 'logFC', term.width = 2)
## Warning: Using size for a discrete variable is not advised.
## Warning: Removed 7 rows containing missing values (geom_point).

层级聚类是一种流行的基因表达无监督聚类分析方法,可确保无偏差的将基因按表达模式组合在一起,因此聚在一起的类可能包含多组共调节或功能相关的基因。GOCluster使用核心R中的hclust方法执行基因表达谱的层级聚类。如果要更改距离度量或聚类算法,请分别使用参数metric和clust,得到的树形图可在ggdendro的帮助下进行转换,并能用ggplot2进行可视化。选择圆形布局,因为它不仅有效而且视觉上吸引人。树形图旁边的第一个圆环代表基因的logFC,它实际上是聚类树的叶子。如果您对多个对比感兴趣,可以修改nlfc参数,默认情况下,它设置为“1”,因此只绘制一个环。logFC值使用用户可定义的色标(lfc.col)进行颜色编码;下一个圆环表示分配给基因的通路。为了好看,对通路数目进行了削减,通路的颜色可以使用参数term.col来。依然可以使用?GOCluster来查看如何更改参数。这个函数最重要的参数是clust.by,可以指定它用基因表达模式(‘logFC’,如上图)或功能类别(‘terms’)进行聚类。
GOCluster(circ, EC$process, clust.by = 'term', lfc.col = c('darkgoldenrod1', 'black', 'cyan1'))
## Warning: Using size for a discrete variable is not advised.
## Warning: Removed 7 rows containing missing values (geom_point).

GOVenn–维恩图
维恩图可用于检测各种差异表达基因列表之间的关系,或探索功能分析中多个通路基因的交集。维恩图不仅显示重叠基因的数量,还显示有关基因表达模式的信息(通常是上调,通常是下调或反调节)。目前,最多三个数据集作为输入。输入数据至少包含两列:一列用于基因名称,一列用于logFC值。
l1 <- subset(circ, term == 'heart development', c(genes,logFC))
l2 <- subset(circ, term == 'plasma membrane', c(genes,logFC))
l3 <- subset(circ, term == 'tissue morphogenesis', c(genes,logFC))
GOVenn(l1,l2,l3, label = c('heart development', 'plasma membrane', 'tissue morphogenesis'))
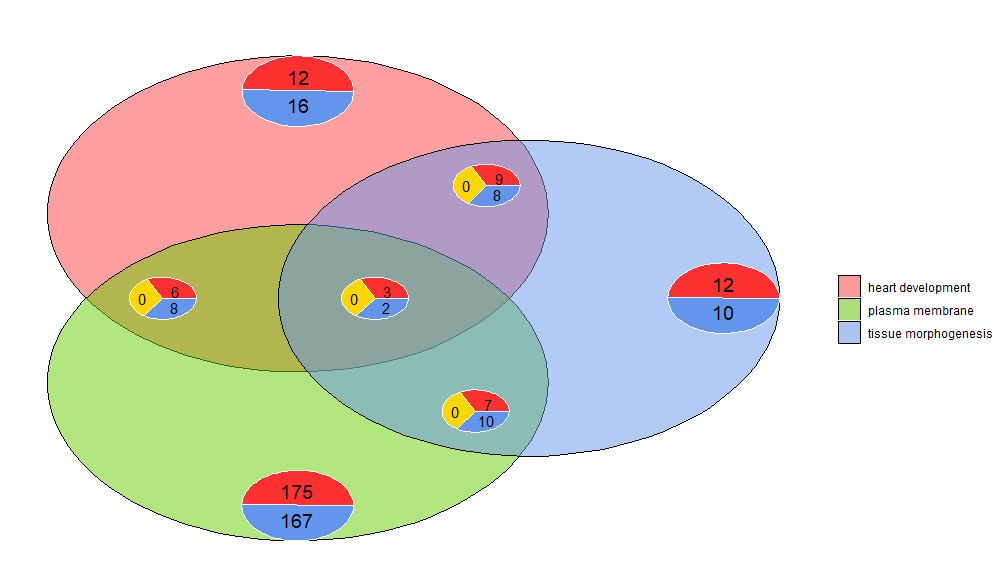
例如,心脏发育和组织形态发生有22个基因,12个是上调的,10个是下调的。需要注意的重要一点是,饼图不显示冗余信息。因此,如果比较三个数据集,则所有数据集共有的基因(中间的饼图)不包含在其他饼图中。可使用此工具的shinyapp https://wwalter.shinyapps.io/Venn/, Web工具更具交互性,圆与数据集的基因数量成面积比例,并且可以使用滑块移动小饼图,并且具有GOVenn功能的所有选项来改变图的布局,也可以下载图片和基因列表。
软件主页:https://wencke.github.io/