Signal Transducers and Activators of Transcription-1 (STAT1) Regulates microRNA Transcription in
Signal Transducers and Activators of Transcription-1(STAT1) Regulates microRNA Transcription in Interferon c-Stimulated HeLa Cells
部分翻译
结论
为了了解在γ干扰素刺激下的海拉细胞内以小RNA为介导的调控网络作用,我们对RNA聚合酶II和STAT1的芯片测序数据进行了生物信息学方法分析,这些芯片测序数据是由CAMDA2009 数据集[11]提供的。我们的分析方法分三个步骤:(1)确定海拉细胞内基因间小RNA的启动子调控区域;(2)确定与STAT1协同作用的转录协同因子;(3)构建在小RNA介导的细胞应答反应中的潜在调控网络基序。
确定基因间小RNA的启动子调控区域
在这个研究中,我们用CAMDA2009数据集[11]提供的RNA聚合酶II(RPlo II)和STAT1的芯片测序数据来确定海拉细胞中基因间小RNA的启动子区域。假定RNA聚合酶II在转录起始位点(TSS)附近的结合位置分布状况,对于转录小RNA的基因和对于编码蛋白质的基因来说都是相似的,我们的工作流程包括三部分[22]:1)对高表达的蛋白质编码基因构建RNA聚合酶II在转录起始位点附近的结合分布模型,2)评估模型性能,3)根据建立的模型在已有注释的小RNA上游区域预测启动子区域。在第一步中,高表达基因是基于在Affymetrix平台上的芯片实验数据选择的。(GEO 序列号:GSE3051 [23])。接下来我们用了一个类似于早前使用过的方法[22],我们只关注那些转录长度大于10000bp,并且在它们的TSS附近10000bp内没有其他基因出现的基因。通过使用5.0版的Affymetrix Microarray Suite平台,基于absent and present calls数据,这种分析方法在海拉细胞中确定出了4120个表达基因和2682个非表达基因。为了评估我们的模型使用RPol II 芯片测序数据预测海拉细胞中活跃启动子的能力,我们随即选取了1/4的表达基因来训练我们的模型。我们把剩下的基因,包括表达的和非表达的,都用作测试数据集。如图一所示,在区分所有的表达基因和非表达基因方面,AUC在ROC曲线中达到了0.86,这表明我们的模型具有很好预测能力。基于基因的表达水平,我们进一步将表达基因分为三类:低表达水平,中表达水平和高表达水平,每一类别包含相同数量的基因。图一的分析结果清晰的证明了我们的模型对于高表达基因具有较高的预测准确性。
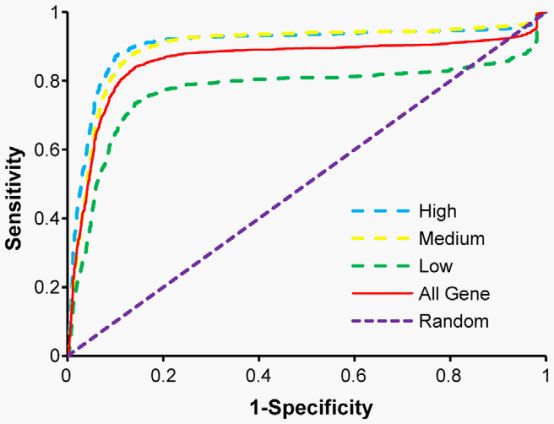
图1.预测蛋白质编码基因的转录起始位点的ROC曲线。表达基因被分为三部分:高表达基因(亮蓝色),低表达基因(绿色),中表达基因(黄色)。表达基因和非表达基因分出来的这三类相应的被认为成正负集合。四分之一的基因被用作训练数据集,剩下的被用作测试数据集。ROC曲线是用R程序中的ROCR库生成的。(http://www.r-project.org) doi:10.1371/journal.pone.0011794.g001
我们在miRBase 小RNA序列数据库中得到了685个人类的成熟小RNA或者小RNA前体的注释数据。(11.0 版本,[3])。其中419个基因间小RNA(位于两个蛋白质编码基因之间)用于启动子预测。使用基于RPol II在蛋白质编码基因转录起始位点附近结合模式所建立的模型参数,我们在海拉细胞内确定了83个活跃的小RNA启动子(错误识别率≤0.2,详见表S1)。调控区间长度的中位数等于1476bp,最长和最短区间长度各自为1476bp和397bp(图2A)。这些区间被认为位于小RNA前体(pri-microRNA)转录起始位点周围,这些小RNA前体长度大约为几百到几千个核苷酸,并且包含一个或者多个小RNA茎环;pri-microRNAs 加工之后形成 pre-microRNA,之后才形成成熟的小RNA形式(microRNA)[25,26]。从确定的TSS到成熟 microRNA或者是pre-microRNA之间的距离也差别很大,分布在200—10000bp之间,中位数距离为3600bp(图2B)。
我们进一步检测了特定的启动子区域的序列特征,包括它们在进化中的保守性,还有与带注释的CpG岛的关系。我们观察到在预测的调控区域内或区域周围有较高的GC含量。在83个预测的microRNA启动子中,有66个启动子(79.5%)被发现含有或者重复含有CpG岛[27]。因为P值<10-77,所以这个结果是十分明显的;这个P值是基于10000次排列计算出来的,这些排列用来评估从83个启动子区域中随机选出66个包含或重复包含CpG岛的概率。另外,这些特定的启动子区域和转录起始位点相对于随机选取的区域(图3中红色虚线)也显示出了较高的保守性(基于17个物种的OgastCons得分,包括哺乳类、两栖类、鸟类、鱼类[27])。
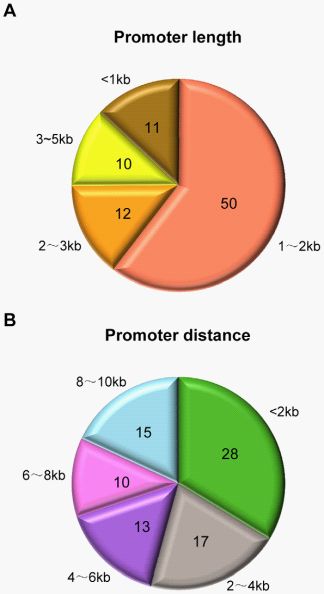
图2.预测的microRNA启动子统计分析。饼形图A显示了不同启动子区间长度,饼形图B显示了不同启动子从预测的转录起始位点到成熟microRNA或pre-microRNA的距离。doi:10.1371/journal.pone.0011794.g002
确定结合在STAT1结合区域附近的协同转录因子
干扰素刺激可能从以下三个机制上影响STAT1的结合:(1)γ干扰素处理影响了STAT1在海拉细胞中的结合力度,因而潜在的改变了STAT1在全基因组范围内的结合模式;(2)干扰素处理会招募一些其他的转录因子作用于STAT1,从而使得STAT1通过蛋白质—蛋白质相互作用与DNA交流;(3)γ干扰素改变了结合在STAT1附近的协同转录因子与DNA的结合活动。为了探索三种潜在机制的可能性,我们在TRANFAC数据库[28]中引进了741个生物学认证的转录结合位点,并浏览了基于ChIp-seq 数据的STAT1 结合区间。像之前叙述过的那样[29],我们用位置特异性得分矩阵来计算一个转录因子在基因组特定位点的结合概率。
。。。。。。。。。。。。。。。。。此处省略N字。。。。。。。。。。。。。。。。。。。。。。。
构建microRNA介导的细胞应答调控网络基序
基于STAT1和它的协同因子在microRNA转录中的转录作用,我们可以导出描述microRNA介导的细胞应答潜在的调控网络元素。在这个研究中,我们对两种网络基序比较感兴趣——反馈和前馈循环,反馈关系描述了microRNA在调控STAT1和它的协同转录因子方面的作用,而前馈关系反映了STAT1调控的microRNA在与STAT1结合的mRNA转录后抑制方面的作用。
反馈回路。图5A显示出了所有的反馈关系体制。这种体制包括两个主要部分,一个之包含STAT1调控,另一个包含STAT1与它的协同调控因子AP1共同调控。从芯片测序数据中,我们可以看到STAT1 在JUN和FOS基因转录起始位点附近结合位点增多,JUN和FOS是与AP-1相结合的转录因子。因此,在γ干扰素的刺激下,STAT1可能刺激JUN和/或FOS,并且,STAT1和JUN/FOS协同调控microRNA表达。
miR-607, miR-92a-1, miR-92b,miR-505这四个microRNA的启动子区域包围着STAT1增多区域或者与之重叠;它们也可能结合到γ干扰素基因的3'不翻译端(图5Bi)。从而通过STAT1-microRNA 介导网络减弱干扰素影响,形成一个反馈关系。类似的关系还存在在miR-1304,miR-24-2, miR-27a, and miR-220c当中。例如,miR-24-2,miR-220c,miR-27a还有miR-1304的预测启动子区域都和一种STAT1的芯片测序区间重叠,这个区间包括STAT1和AP-1的结合位点。这表明这些microRNA是被STAT1与AP-1结合蛋白协同调控的。重要的是,miR-24-2可以反过来结合到JUN基因,FOS基因,γ干扰素基因的3'不翻译区(如图5Bii),因此可能抑制这些基因的mRNA表达水平。类似,miR-220c与STAT1和γ干扰素都能结合(如图5Biii);miR-27a可与STAT1结合(如图5Biv),miR-1304可与FOS基因结合(如图5Bv)。这种反馈关系可能代表了一种microRNA介导的分子机制,这种分子机制使得海拉细胞在受到γ干扰素处理时保持内稳态。
前馈关系。这种前馈关系反映了STAT1调控的microRNA在与STAT1结合的mRNA转录后抑制方面的作用。既然STAT1可能在靶基因上发挥刺激作用或者抑制作用,那么它也可能代表了海拉细胞调控STAT1诱导活化反应的一种机制。这种关系也叫做“无逻辑前馈调控”[30],也被Marson等人发现过[10]。在STAT1增多区域围绕或者包含他们调控区域(从转录起始位点的-1000bp到+500bp)的6264个基因中,有1265个基因(20.2%)被预测为37个与STAT1结合的microRNA的靶基因(图6)。补充材料的表S4给出了一系列假定的前馈关系。
author:大海浪涛