什么是高通量测序?
高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十万到几百万条核酸分子进行序列测定, 因此在有些文献中称其为下一代测序技术(next generation sequencing,NGS )足见其划时代的改变, 同时高通量测序使得对一个物种的转录组和基因组进行细致全貌的分析成为可能, 所以又被称为深度测序(Deep sequencing)。
什么是Sanger法测序(一代测序)
Sanger法测序利用一种DNA聚合酶来延伸结合在待定序列模板上的引物。直到掺入一种链终止核苷酸为止。每一次序列测定由一套四个单独的反应构成,每个反应含有所有四种脱氧核苷酸三磷酸(dNTP),并混入限量的一种不同的双脱氧核苷三磷酸(ddNTP)。由于ddNTP缺乏延伸所需要的3-OH基团,使延长的寡聚核苷酸选择性地在G、A、T或C处终止。终止点由反应中相应的双脱氧而定。每一种dNTPs和ddNTPs的相对浓度可以调整,使反应得到一组长几百至几千碱基的链终止产物。它们具有共同的起始点,但终止在不同的的核苷酸上,可通过高分辨率变性凝胶电泳分离大小不同的片段,凝胶处理后可用X-光胶片放射自显影或非同位素标记进行检测。
什么是基因组重测序(Genome Re-sequencing)
全基因组重测序是对基因组序列已知的个体进行基因组测序,并在个体或群体水平上进行差异性分析的方法。随着基因组测序成本的不断降低,人类疾病的致病突变研究由外显子区域扩大到全基因组范围。通过构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,实现在全基因组水平上检测疾病关联的常见、低频、甚至是罕见的突变位点,以及结构变异等,具有重大的科研和产业价值。
什么是de novo测序
de novo测序也称为从头测序:其不需要任何现有的序列资料就可以对某个物种进行测序,利用生物信息(bioinformation)学分析手段对序列进行拼接,组装,从而获得该物种的基因组图谱。获得一个物种的全基因组序列是加快对此物种了解的重要捷径。随着新一代测序技术的飞速发展,基因组测序所需的成本和时间较传统技术都大大降低,大规模基因组测序渐入佳境,基因组学研究也迎来新的发展契机和革命性突破。利用新一代高通量、高效率测序技术以及强大的生物信息(bioinformation)分析能力,可以高效、低成本地测定并分析所有生物的基因组序列。
什么是外显子测序(whole exon sequencing)
外显子组测序是指利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、Indel等具有较大的优势,但无法研究基因组结构变异如染色体断裂重组等。
什么是mRNA测序 (RNA-seq)
转录组学(transcriptomics)是在基因组学后新兴的一门学科,即研究特定细胞在某一功能状态下所能转录出来的所有RNA(包括mRNA和非编码RNA)的类型与拷贝数。Illumina提供的mRNA测序技术可在整个mRNA领域进行各种相关研究和新的发现。mRNA测序不对引物或探针进行设计,可自由提供关于转录的客观和权威信息。研究人员仅需要一次试验即可快速生成完整的poly-A尾的RNA完整序列信息,并分析基因表达、cSNP、全新的转录、全新异构体、剪接位点、等位基因特异性表达和罕见转录等最全面的转录组信息。简单的样品制备和数据分析软件支持在所有物种中的mRNA测序研究。
什么是small RNA测序
Small RNA(micro RNAs、siRNAs和 pi RNAs)是生命活动重要的调控因子,在基因表达调控、生物个体发育、代谢及疾病的发生等生理过程中起着重要的作用。Illumina能够对细胞或者组织中的全部Small RNA进行深度测序及定量分析等研究。实验时首先将18-30 nt范围的Small RNA从总RNA中分离出来,两端分别加上特定接头后体外反转录做成cDNA再做进一步处理后,利用测序仪对DNA片段进行单向末端直接测序。通过Illumina对Small RNA大规模测序(Large-scale sequencing)分析,可以从中获得物种全基因组水平的miRNA图谱,实现包括新miRNA分子的挖掘,其作用靶基因的预测和鉴定、样品间差异表达分析、miRNAs聚类和表达谱分析等科学应用。
什么是miRNA测序
成熟的microRNA(miRNA)是17~24nt的单链非编码RNA分子,通过与mRNA相互作用影响目标mRNA的稳定性及翻译,最终诱导基因沉默,调控着基因表达、细胞生长、发育等生物学过程。基于第二代测序技术的microRNA测序,可以一次性获得数百万条microRNA序列,能够快速鉴定出不同组织、不同发育阶段、不同疾病状态下已知和未知的microRNA及其表达差异,为研究microRNA对细胞进程的作用及其生物学影响提供了有力工具。
什么是Chip-seq
染色质免疫共沉淀技术(ChromatinImmunoprecipitation,ChIP)也称结合位点分析法,是研究体内蛋白质与DNA相互作用的有力工具,通常用于转录因子结合位点或组蛋白特异性修饰位点的研究。将ChIP与第二代测序技术相结合的ChIP-Seq技术,能够高效地在全基因组范围内检测与组蛋白、转录因子等互作的DNA区段。
ChIP-Seq的原理是:首先通过染色质免疫共沉淀技术(ChIP)特异性地富集目的蛋白结合的DNA片段,并对其进行纯化与文库构建;然后对富集得到的DNA片段进行高通量测序。研究人员通过将获得的数百万条序列标签精确定位到基因组上,从而获得全基因组范围内与组蛋白、转录因子等互作的DNA区段信息。
什么是CHIRP-Seq
CHIRP-Seq( Chromatin Isolation by RNA Purification )是一种检测与RNA绑定的DNA和蛋白的高通量测序方法。方法是通过设计生物素或链霉亲和素探针,把目标RNA拉下来以后,与其共同作用的DNA染色体片段就会附在到磁珠上,最后把染色体片段做高通量测序,这样会得到该RNA能够结合到在基因组的哪些区域,但由于蛋白测序技术不够成熟,无法知道与该RNA结合的蛋白。
什么是RIP-seq
RNA Immunoprecipitation是研究细胞内RNA与蛋白结合情况的技术,是了解转录后调控网络动态过程的有力工具,能帮助我们发现miRNA的调节靶点。这种技术运用针对目标蛋白的抗体把相应的RNA-蛋白复合物沉淀下来,然后经过分离纯化就可以对结合在复合物上的RNA进行测序分析。
RIP可以看成是普遍使用的染色质免疫沉淀ChIP技术的类似应用,但由于研究对象是RNA-蛋白复合物而不是DNA-蛋白复合物,RIP实验的优化条件与ChIP实验不太相同(如复合物不需要固定,RIP反应体系中的试剂和抗体绝对不能含有RNA酶,抗体需经RIP实验验证等等)。RIP技术下游结合microarray技术被称为RIP-Chip,帮助我们更高通量地了解癌症(cancer)以及其它疾病整体水平的RNA变化。
什么是CLIP-seq
CLIP-seq,又称为HITS-CLIP,即紫外交联免疫沉淀结合高通量测序(crosslinking-immunprecipitation and high-throughput sequencing), 是一项在全基因组水平揭示RNA分子与RNA结合蛋白相互作用的革命性技术。其主要原理是基于RNA分子与RNA结合蛋白在紫外照射下发生耦联,以RNA结合蛋白的特异性抗体将RNA-蛋白质复合体沉淀之后,回收其中的RNA片段,经添加接头、RT-PCR等步骤,对这些分子进行高通量测序,再经生物信息(bioinformation)学的分析和处理、总结,挖掘出其特定规律,从而深入揭示RNA结合蛋白与RNA分子的调控作用及其对生命的意义。
什么是metagenomic(宏基因组)
Magenomics研究的对象是整个微生物群落。相对于传统单个细菌研究来说,它具有众多优势,其中很重要的两点:(1) 微网络常是以群落方式共生于某一小生境中,它们的很多特性是基于整个群落环境及个体间的相互影响的,因此做Metagenomics研究比做单个个体的研究更能发现其特性;(2) Metagenomics研究无需分离单个细菌,可以研究那些不能被实验室分离培养的微生物。
宏基因组是基因组学一个新兴的科学研究方向。宏基因组学(又称元基因组学,环境基因组学,生态基因组学等),是研究直接从环境样本中提取的基因组遗传物质的学科。传统的微生物研究依赖于实验室培养,元基因组的兴起填补了无法在传统实验室中培养的微生物研究的空白。过去几年中,DNA测序技术的进步以及测序通量和分析方法的改进使得人们得以一窥这一未知的基因组科学领域。
什么是SNP、SNV(单核苷酸位点变异)
单核苷酸多态性singlenucleotide polymorphism,SNP 或单核苷酸位点变异SNV。个体间基因组DNA序列同一位置单个核苷酸变异(替代、插入或缺失)所引起的多态性。不同物种、个体基因组DNA序列同一位置上的单个核苷酸存在差别的现象。有这种差别的基因座、DNA序列等可作为基因组作图的标志。人基因组上平均约每1000个核苷酸即可能出现1个单核苷酸多态性的变化,其中有些单核苷酸多态性可能与疾病有关,但可能大多数与疾病无关。单核苷酸多态性是研究人类家族和动植物品系遗传变异的重要依据。在研究癌症(cancer)基因组变异时,相对于正常组织,癌症(cancer)中特异的单核苷酸变异是一种体细胞突变(somatic mutation),称做SNV。
什么是INDEL (基因组小片段插入)
基因组上小片段(>50bp)的插入或缺失,形同SNP/SNV。
什么是copy number variation(****CNV****):基因组拷贝数变异
基因组拷贝数变异是基因组变异的一种形式,通常使基因组中大片段的DNA形成非正常的拷贝数量。例如人类正常染色体拷贝数是2,有些染色体区域拷贝数变成1或3,这样,该区域发生拷贝数缺失或增加,位于该区域内的基因表达量也会受到影响。如果把一条染色体分成A-B-C-D四个区域,则A-B-C-C-D/A-C-B-C-D/A-C-C-B-C-D/A-B-D分别发生了C区域的扩增及缺失,扩增的位置可以是连续扩增如A-B-C-C-D也可以是在其他位置的扩增,如A-C-B-C-D。
什么是structure variation(SV):基因组结构变异
染色体结构变异是指在染色体上发生了大片段的变异。主要包括染色体大片段的插入和缺失(引起CNV的变化),染色体内部的某块区域发生翻转颠换,两条染色体之间发生重组(inter-chromosome trans-location)等。一般SV的展示利用Circos 软件。
什么是Segment duplication
一般称为SD区域,串联重复是由序列相近的一些DNA片段串联组成。串联重复在人类基因多样性的灵长类基因中发挥重要作用。在人类染色体Y和22号染色体上,有很大的SD序列。
什么是genotype and phenotype**
既基因型与表型;一般指某些单核苷酸位点变异与表现形式间的关系。
什么 Read Contig Unigene
高通量测序时,在芯片上的每个反应,会读出一条序列,是比较短的,叫read,它们是原始数据;
有很多reads通过片段重叠,能够组装成一个更大的片段,称为contig;
多个contigs通过片段重叠,组成一个更长的scaffold;
一个contig被组成出来之后,鉴定发现它是编码蛋白质的基因,就叫singleton;
多个contigs组装成scaffold之后,鉴定发现它编码蛋白质的基因,叫unigene。
**UniGene
UniGene是以自动化的方式,对于每一个新进入到GeneBank的序列,进行序列相似性分析,如果可以找到可能是来自于同一个基因的基因组(cluster),则将次序列归入到这一个基因组,如果找不到,则成立一个新的基因组。据估计,人类的基因约有八万到十万个左右,而在UniGenes中的所有人类序列中,经过上述方式加以分组之后,在1998您6月,已得到的超过四万三千个独特的基因组(unique gene clusters),其中大约六千余个具有已知的基因。
什么是soft-clipped reads
当基因组发生某一段的缺失,或转录组的剪接,在测序过程中,横跨缺失位点及剪接位点的reads回帖到基因组时,一条reads被切成两段,匹配到不同的区域,这样的reads叫做soft-clipped reads,这些reads对于鉴定染色体结构变异及外源序列整合具有重要作用。
什么是multi-hits reads
由于大部分测序得到的reads较短,一个reads能够匹配到基因组多个位置,无法区分其真实来源的位置。一些工具根据统计模型,如将这类reads分配给reads较多的区域。
什么是Scaffold
基因组de novo测序,通过reads拼接获得Contigs后,往往还需要构建454 Paired-end库或Illumina Mate-pair库,以获得一定大小片段(如3Kb、6Kb、10Kb、20Kb)两端的序列。基于这些序列,可以确定一些Contig之间的顺序关系,这些先后顺序已知的Contigs组成Scaffold。
什么是Contig N50
Reads拼接后会获得一些不同长度的Contigs。将所有的Contig长度相加,能获得一个Contig总长度。然后将所有的Contigs按照从长到短进行排序,如获得Contig 1,Contig 2,Contig 3...………Contig 25。将Contig按照这个顺序依次相加,当相加的长度达到Contig总长度的一半时,最后一个加上的Contig长度即为Contig N50。举例:Contig 1+Contig 2+ Contig 3+Contig 4=Contig总长度*1/2时,Contig 4的长度即为Contig N50。Contig N50可以作为基因组拼接的结果好坏的一个判断标准。
什么是Scaffold N50
Scaffold N50与Contig N50的定义类似。Contigs拼接组装获得一些不同长度的Scaffolds。将所有的Scaffold长度相加,能获得一个Scaffold总长度。然后将所有的Scaffolds按照从长到短进行排序,如获得Scaffold 1,Scaffold 2,Scaffold 3...………Scaffold 25。将Scaffold按照这个顺序依次相加,当相加的长度达到Scaffold总长度的一半时,最后一个加上的Scaffold长度即为Scaffold N50。举例:Scaffold 1+Scaffold 2+ Scaffold 3 +Scaffold 4 +Scaffold 5=Scaffold总长度*1/2时,Scaffold 5的长度即为Scaffold N50。Scaffold N50可以作为基因组拼接的结果好坏的一个判断标准。
什么是测序深度和覆盖度
测序深度(Sequencing Depth):测序得到的碱基总量(bp)与基因组大小(Genome)的比值,它是评价测序量的指标之一。测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。重测序的个体,如果采用的是双末端或Mate-Pair方案,当测序深度在10~15X以上时,基因组覆盖度和测序错误率控制均得以保证。
假设一个基因大小为2M,测序深度为10X,那么获得的总数据量为20M。覆盖度是指测序获得的序列占整个基因组的比例。由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖有所的区域,这部分没有获得的区域就称为Gap。例如一个细菌基因组测序,覆盖度是98%,那么还有2%的序列区域是没有通过测序获得的。
denovo字面意思是全新,专业一点就是从头测序。详细点就是对未知基因组序列进行测序,利用生物信息(bioinformation)学分析手段,对序列进行拼接、组装,从而获得其基因组的图谱。
测序的覆盖度(coverage)和测序的深度(depth)。对于coverage,由于大片段拼接的gap(空白或者缺口)、测序读长有限、重复序列等问题的存在,测序分析后组装得到的基因组序列通常无法完全覆盖所有区域,覆盖度就是最终得到的结果占整个基因组的比例。例如一个人的基因组测序,覆盖度为98.5%,那么说明该基因组还有1.5%的区域通过我们的组装和分析无法得到;对于depth,就是被测基因组上单个碱基被测序的平均次数,比如某样本的测序深度为30X,那么就是说该样本的基因组上每一个单碱基平均被测序(或者说读取)了30次,注意,是平均。当然了,depth也有最大和最小值,这个都可以由信息分析得到。其实也就是为了提高准确率什么的,一般15X就差不多了。
什么是DeBruijn图
Kautz和DeBruijn图由于其在大型计算机互联网上的应用而被人们广泛的研究,互联网的一个重要的参数是它的等周数.Deplormc和TiⅡich运用特征值技术发现了Kautz和De-Bruijn图等周数的一个上界.Buherman给出了一个构造性的方法改进了DeBruijn图等周数的上).我们运用该构造方法得到了Kautz图的一个新的上界.什么是RPKM、FPKM
RPKM,Reads Per Kilobase of exon model per Million mapped reads, is defined in thisway [Mortazavi etal., 2008]:
每1百万个map上的reads中map到外显子的每1K个碱基上的reads个数。是将map到基因的read数除以map到genome的所有read数(以million为单位)与RNA的长度(以KB为单位)。
RNA-seq是透过次世代定序的技术来侦测基因表现量的方法,在衡量基因表现量时,若是单纯以map到的read数来计算基因的表现量,在统计上是一件相当不合理事,因为在随机抽样的情况下,序列较长的基因被抽到的机率本来就会比序列短的基因较高,如此一来,序列长的基因永远会被认为表现量较高,而错估基因真正的表现量,所以Ali Mortazavi等人在2008年提出以RPKM在估计基因的表现量

举例:比如对应到该基因的read有1000个,总reads个数有100万,而该基因的外显子总长为5kb,那么它的RPKM为:109*1000(reads个数)/106(总reads个数)5000(外显子长度)=200或者:1000(reads个数)/1(百万)5(K)=200这个值反映基因的表达水平。
FPKM(fragments per kilobase of exon per million fragments mapped).每1百万个map上j的reads中map到外显子的每1K个碱基上的reads个数。FPKM与RPKM计算方法基本一致。不同点就是FPKM计算的是fragments,而RPKM计算的是reads。Fragment比read的含义更广,因此FPKM包含的意义也更广,可以是pair-end的一个fragment,也可以是一个read。

什么是转录本重构
用测序的数据组装成转录本。有两种组装方式:1,de-novo构建; 2,有参考基因组重构。其中de-novo组装是指在不依赖参考基因组的情况下,将有overlap的reads连接成一个更长的序列,经过不断的延伸,拼成一个个的contig及scaffold。常用工具包括velvet,trans-ABYSS,Trinity等。有参考基因组重构,是指先将read贴回到基因组上,然后在基因组通过reads覆盖度,junction位点的信息等得到转录本,常用工具包括scripture、cufflinks。
什么是genefusion
将基因组位置不同的两个基因中的一部分或全部整合到一起,形成新的基因,称作融合基因,或嵌合体基因。该基因有可能翻译出融合或嵌合体蛋白。
什么是表达谱
基因表达谱(geneexpression pro
file):指通过构建处于某一特定状态下的细胞或组织的非偏性cDNA文库,大规模cDNA测序,收集cDNA序列片段、定性、定量分析其mRNA群体组成,从而描绘该特定细胞或组织在特定状态下的基因表达种类和丰度信息,这样编制成的数据表就称为基因表达谱 什么是功能基因组学(functional genomics)
功能基因组学(functional genomics)(Functuionalgenomics)又往往被称为后基因组学(Postgenomics),它利用结构基因组所提供的信息和产物,发展和应用新的实验手段,通过在基因组或系统水平上全面分析基因的功能,使得生物学研究从对单一基因或蛋白质得研究转向多个基因或蛋白质同时进行系统的研究。这是在基因组静态的碱基序列弄清楚之后转入对基因组动态的生物学功能学研究。研究内容包括基因功能发现、基因表达分析及突变检测。基因的功能包括:生物学功能,如作为蛋白质激酶对特异蛋白质进行磷酸化修饰;细胞学功能,如参与细胞间和细胞内信号传递途径;发育上功能,如参与形态建成等。采用的手段包括经典的减法杂交,差示筛选,cDNA代表差异分析以及mRNA差异显示等,但这些技术不能对基因进行全面系统的分析,新的技术应运而生,包括基因表达的系统分析(serial analysis of gene expr
ession,SAGE),cDNA微阵列(cDNA microarray),DNA 芯片(DNA chip)和序列标志片段显示(sequence tagged fragmentsdisplay。 什么是比较基因组学(comparative genomics)
比较基因组学(comparative genomics)(ComparativeGenomics)是基于基因组图谱和测序基础上,对已知的基因和基因组结构进行比较,来了解基因的功能、表达机理和物种进化的学科。利用模式生物(model organism)基因组与人类基因组之间编码顺序上和结构上的同源性,克隆人类疾病基因,揭示基因功能和疾病分子机制(Molecular Mechanisms),阐明物种进化关系,及基因组的内在结构。
什么是表观遗传学
表观遗传学是研究基因的核苷酸序列不发生改变的情况下,基因表达了可遗传的变化的一门遗传学分支学科。表观遗传的现象很多,已知的有DNA甲基化(DNAmethylation),基因组印记(genomicimpriting),母体效应(maternaleffects),基因沉默(genesilencing),核仁显性,休眠转座子激活和RNA编辑(RNA editing)等。
什么是计算生物学
计算生物学是指开发和应用数据分析及理论的方法、数学建模、计算机仿真技术等。当前,生物学数据量和复杂性不断增长,每14个月基因研究产生的数据就会翻一番,单单依靠观察和实验已难以应付。因此,必须依靠大规模计算模拟技术,从海量信息中提取最有用的数据。
什么是基因组印记
基因组印记(又称遗传印记)是指基因根据亲代的不同而有不同的表达。印记基因的存在能导致细胞中两个等位基因的一个表达而另一个不表达。基因组印记是一正常过程,此现象在一些低等动物和植物中已发现多年。印记的基因只占人类基因组中的少数,可能不超过5%,但在胎儿的生长和行为发育中起着至关重要的作用。基因组印记病主要表现为过度生长、生长迟缓、智力障碍、行为异常。目前在肿瘤的研究中认为印记缺失是引起肿瘤最常见的遗传学因素之一。
什么是基因组学
基因组学(英文genomics),研究生物基因组和如何利用基因的一门学问。用于概括涉及基因作图、测序和整个基因组功能分析的遗传学分支。该学科提供基因组信息以及相关数据系统利用,试图解决生物,医学,和工业领域的重大问题。
什么是DNA甲基化
CpG岛,英文名称:CpG island
定义:位于多种脊椎动物(vertebrates)已知基因转录起始位点周围、由胞嘧啶(C)和鸟嘧啶(G)组成的串联重复序列。
CpG岛(CpG island):CpG双核苷酸在人类基因组中的分布很不均一,而在基因组的某些区段,CpG保持或高于正常概率,这些区段被称作CpG岛
DNA甲基化是指在DNA甲基化转移酶的作用下,在基因组CpG二核苷酸的胞嘧啶5"碳位共价键结合一个甲基基团。正常情况下,人类基因组“垃圾”序列的CpG二核苷酸相对稀少,并且总是处于甲基化状态,与之相反,人类基因组中大小为100—1000 bp左右且富含CpG二核苷酸的CpG岛则总是处于未甲基化状态,并且与56%的人类基因组编码基因相关。人类基因组序列草图分析结果表明,人类基因组CpG岛约为28890个,大部分染色体每1 Mb就有5—15个CpG岛,平均值为每Mb含10.5个CpG岛,CpG岛的数目与基因密度有良好的对应关系[9]。由于DNA甲基化与人类发育和肿瘤疾病的密切关系,特别是CpG岛甲基化所致抑癌基因转录失活问题,DNA甲基化已经成为表观遗传学和表观基因组学的重要研究内容。
什么是基因组注释
基因组注释(Genomeannotation) 是利用生物信息(bioinformation)学方法和工具,对基因组所有基因的生物学功能进行高通量注释,是当前功能基因组学(functional genomics)研究的一个热点。基因组注释的研究内容包括基因识别和基因功能注释两个方面。基因识别的核心是确定全基因组序列中所有基因的确切位置。
以上转自http://www.plob.org/2012/11/21/4810.html,稍加修改。
E-value
EXPECT
E期望值(E-value)这个数值表示你仅仅因为随机性造成获得这一alignment结果的可能次数。这一数值越接近零,发生这一事件的可能性越小。从搜索的角度看,E值越小,alignment结果越显著。你可能会想为搜索设定一个期望值阀值(EXPECT),例如Defaults值设为10。这一设置则表示联配结果中将有10个匹配序列是由随机产生,如果联配的统计显著性值(E值)小于该值(10),则该alignment将被检出,换句话说,比较低的阀值将使搜索的匹配要求更严格,结果报告中随机产生的匹配序列减少。E=kmne^(-λs)
RNA Integrity Number (RIN)
The RNA integrity number (RIN) is a software tool designed to help scientists estimate the integrity of total RNA samples
TRS、DRS、SSR
根据重复序列在基因组中的分布形式可将其分为串联重复序列(Tandem Repeats Sequence,TRS)和散布重复序列(Dispersed Repeats Sequence,DRS)。其中,串联重复序列是由相关的重复单位首尾相连、成串排列而成的。发现的串联重复序列主要有两类:一类是由功能基因组成的(如rRNA和组蛋白基因);另一类是由无功能的序列组成的。
根据重复序列的重复单位的长度,可将串联重复序列分为卫星DNA、微卫星DNA、小卫星 DNA等。微卫星DNA又叫简单重复序列(Simple Sequence Repeat,SSR),指的是基因组中由1-6个核苷酸组成的基本单位重复多次构成的一段DNA,广泛分布于基因组的不同位置,长度一般在200 bp以下。
简单重复序(SSR)也称微卫星DNA,其串联重复的核心序列为1一6 bp,其中最常见是双核苷酸重复,即(CA) n和(TG) n每个微卫星DNA的核心序列结构相同,重复单位数目10一60个,其高度多态性主要来源于串联数目的不同。
根据SSR核心序列排列方式的不同,可分为3种类型:
完全型(perfect)。指核心序列以不间断的重复方式首尾相连构成的DNA。如: ATATATATATATATATATATATATATATATATAT
不完全型(imperfect)。指在SSR的核心序列之间有3个以下的非重复碱基,但两端的连续重复核心序列重复数大于3。如:ATATATATGGATATATATATCGATATATATATATATATGGATATATATAT
复合型(compound)。指2个或2个以上的串联核心序列由3个或3个以上的连续的非重复碱基分隔开,但这种连续性的核心序列重复数不少于5。如:ATATATATATATATGGGATATATATATATA
3种类型中完全型是SSR标记中应用较多的一种类型。
结构域(structure domain)是在蛋白质三级结构中介于二级和三级结构之间的可以明显区分但又相对独立的折叠单元,每个结构域自身形成紧实的三维结构,可以独立存在或折叠,但结构域与结构域之间关系较为松散。
结构功能域通常由25~300个氨基酸残基组成,不同蛋白质分子中结构域的数目不同,同一个蛋白质分子中的几个结构域彼此相似或者不尽相同。结构域是蛋白质的功能、结构和进化单位,结构功能域分析对于蛋白质结构的分类和预测有着重要的作用。
motif又称模体,是序列中局部的保守区域,或者是一组序列中共有的一小段序列模式。一般指构成任何一种特征序列的基本结构,但是多数情况下是指可能具有分子功能、结构性质或家族成员相关的任何序列模式。
motif作为结构域中的亚单位,表现结构域的各种生物学功能。常见的蛋白质结构motif,种类超过28类。常见的motif搜索方法主要基于两种,一种是序列模式(Pattern),另外一种是序列特征谱(Pro
file)。 序列模式方法直接搜索关键的几个保守残基,忽略其他位置的氨基酸多态性。例如,“L-x(6)-L-x(6)-L-x(6)-L”(x表示任意氨基酸)为亮氨酸拉链结构的序列模式,这样一段序列多处于蛋白质的活性区域或重要结构区,较为保守,是motif搜索的目标之一。由于序列模式方法搜索的不是完整的结构域或整个蛋白的特征,故其适用于识别保守的功能区域,对于序列变异大的功能区域,则无法准确识别。此外,随机的氨基酸序列也可能出现短小的序列模式,故易产生假阳性,对于此类搜索需要搜索多个不同的数据库,得到尽可能多得同源序列,从而才能更好的说明序列中包含的信息。
序列特征谱搜索是基于蛋白质序列多重比对结果中的保守序列区域进行搜索,由于考虑了不同保守度的氨基酸在相应位置的权重,可以更为敏感的检测到进化距离较远的蛋白质相关性,得到比序列模式方法更为灵敏的结果,但可靠的序列特征谱数目往往有限,因此该方法在进行新基因功能预测时受到了较大的障碍。
Q-1****:覆盖率(Coverage ratio)是什么?覆盖深度(Coverage depth)是什么?测序深度和基因组覆盖率的关系如何?
A-1:覆盖比率,亦简称覆盖率,指被测序到的碱基占全基因组大小的比率。
覆盖深度,亦简称覆盖度,指每个碱基被测序的平均次数。
测序深度指平均碱基测序深度(测序的数据总量比上基因组大小)。测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。重测序的个体,如果采用的是双末端或Mate-Pair方案,当测序深度在10-15X以上时,基因组覆盖度和测序错误率控制均得以保证。
Q-2****:测序完成后的基因组、基因区域覆盖度是多少?
A-2:基因组覆盖度达到95%以上,基因区域覆盖度98%以上。
Q-3****:一致性序列组装和基因组组装有什么不同?
A-3:基因组组装:没有参考基因组,首先需要构建多个不同插入片段长度的双末端文库。根据短reads的overlap关系将其组装成contig,然后利用大片段文库双末端关系和插入片段的大小,将contig又连接成scaffold,每个scaffold可能是由几个contig组成,中间会有gap的存在,然后再进行补洞。如果该物种有比较好的连锁图谱的话,可以将scaffolds定位回染色体。
一致性序列组装:根据与参考序列的比对结果,我们利用贝叶斯模型得到测序个体每个位点可能性最大的基因型,并组装出该个体的一致序列。
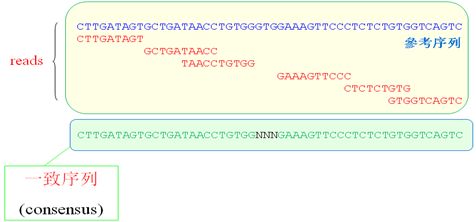
一致序列的组装过程中,只有比对参考基因组具有唯一性的reads才能用于生成一致序列,如上面的示意图没有覆盖到部分用N代替。根据比对结果,综合考虑数据特征、测序质量及实验方面的影响因素,利用贝叶斯模型,在实际观察到的数据基础上计算出每个可能的基因型概率。挑选出概率最大的基因型作为该测序个体的特定位点的基因型,并在此基础上给出一个反映该基因型准确的质量值,并且得到一致序列。可见一致性序列的组装****必须借助于参考基因组,并且只有唯一比对到参考序列上的reads才保留。
Q-4****:重测序都可以检测哪些遗传变异?
A-4:重测序目前能够检测到的遗传变异包括SNP(single nucleotide polymorphism, 单核苷酸多态性)、Indel(Insertion or deletion, 插入或缺失)、SV(structure variation, 结构变异)等。
Q-5****:Indel(insertion or deletion,插入或缺失)的定义?分析软件?如何寻找?
A-5:Indel指的是insertion or deletion,Indel包括插入或缺失突变,或两者兼之。Indel可以作为自然群体特别是进化过程中的遗传标记。
源宜基因基于生成的 bam 文件,使用 GATK 软件识别其中的 SNP 和 InDel 等变异,生成 vcf 格式的变异检测文件。
Q-6:Structure variation****的定义?分析软件?
A-6:染色体发生的结构变异主要有4种:
(1)缺失:染色体中某一片段的缺失。
(2)重复:染色体增加了某一片段。
(3)倒位:染色体某一片段的位置颠倒了180度,造成染色体内的重新排列。
(4)易位:****染色体的某一片段移接到另一条非同源染色体上或同一条染色体上的不同区域。
Q-7****:群体重测序进行信息分析我们能获得什么?
A-7:重测序目前能够检测到遗传变异包括SNP( single nucleotide polymorphism,单核苷酸多态性)、Indel(Insertion or deletion,插入或缺失)、SV(structure variation,结构变异)。除此之外还可以进行连锁不平衡(LD)、系统进化树(phylogenetic tree)、群体遗传结构(Population Genetic Structure)、群体选择分析(Population Selective Analysis)等。
Q-8****:什么是全基因组重测序BSA(WG-BSA)?
A-8:对已有参考基因组序列的所有作图群体(F1, F2, RIL, DH, BC1等),对亲本进行个体重测序,对某个极端性状后代进行混池重测序,检测SNP及Indel等变异位点,通过关联分析精细定位与目标性状相关的基因区域,获得与性状紧密关联的分子标记,并通过功能注释定位到一些候选基因,BSA分析方法是目前最高效的单一性状功能基因定位方法。
Q-9****:全基因组重测序BSA一般最少要求多少样本用于构建混池?
A-9:一般需要20-50个个体。
Q-10****:连锁不平衡(Linkage disequilibrium)是什么?分析软件?
A-10:连锁不平衡(linkage disequilibrium, LD)指的是一个群体内不同座位等位基因之间的非随机关联, 包括两个标记间或两个基因/QTL间或一个基因/QTL与一个标记座位间的非随机关联。连锁不平衡与连锁是相关但完全不同的两个概念。连锁不平衡指的是群体内等位基因之间的相关,而连锁指的是位于同一条染色体上的基因联合传递的现象。紧密连锁可导致较高的LD水平,但这种LD纯粹是由突变产生的等位基因出现后紧密连锁座位间所有重组事件的结果。连锁不平衡分析是近年来的一个研究亮点和热点。基于LD的作图方法不仅是新基因发掘的有效途径,而且也是联系结构基因组学和表型组学的一座桥梁。LD分析软件有Haploview等。
Q-11****:系统进化树是什么?有哪些分析方法或软件?
A-11:系统发生树(phylogenetic tree,又称evolutionary tree进化树)就是描述群体间进化顺序的分支图或树,表示群体间的进化关系。系统分析过程就是指构建群体之间的进化树,推测其亲缘关系的远近。分析软件有MEGA4.0或 PHYLIP 3.68等。
Q-12****:什么是主成分分析(Principal component analysis)?
A-12:主成分分析(Principal component analysis,PCA)是一种纯数学的运算方法,可以将很多可能相关的变量转变成数量更少的主成分变量。PCA应用到很多学科,在遗传学当中,主要用于聚类分析,它是基于个体基因组SNP差异程度,按照不同性状特征将个体按主成分进行聚类成不同的亚群,同时用于和其它方法做相互验证。
Q-13****:群体遗传结构(Population Genetic Structure)是什么?分析方法或软件有哪些?
A-13:群体遗传结构是指遗传变异在物种或群体中的一种非随机分布,即遗传变异在群体内、群体间的分布样式以及在时间上的变化。遗传结构的模式提供了对进化过程的见解,并帮助确定了物种群体基因型和表型关联的研究。分析软件有Structure、Frappe 以及Admixture等。
Q-14****:群体选择分析是什么?有哪些分析方法或软件?
A-14:选择分析就是筛选那些不同亚群之间(如栽培和野生)之间能够造成这两个亚群在进化上为何分开,以及产生重大差异的一些变异位点,从而扩展到基因层面。****选择分析的方法包括Fst value、Heterozygosity、Tajima’s D value、θπ value、HKA test、GORSS test等。
连锁不平衡(LD)分析、系统进化树(phylogeny tree)分析、主成分分析(PCA)、遗传结构分析和群体选择分析都是基于SNPs进行的分析,寻找群体差异。例如野生群体和家养群体之间的相似或者差异较大的区域,相似非常高的可能说明在这段区域在这个物种中可能相对保守,而差异较大的区域可能就是由于受人工选择或者自然选择的敏感区域,这些区域可能与选择密切相关,可能是野生群体和家养群体产生差异的主要原因。
Q-15****:常染色体和性染色全基因组重测序测序深度的差异问题?
A-15:如果所测样本为女性(XX),那么常染色体和性染色体重测序深度基本没有差异;如果所测样本为男性,因为性染色为 XY,因此在计算测序深度时是分开计算,其深度基本只有常染色体的一半;但由于 X 染色体和 Y 染色体同源性比较高,X 和 Y之间也会出现差异较大的情况。
Q-16****:选择性消除分析的常用算法及软件有哪些?
A-16:选择消除分析是通过比较不同群体的差异,分析各亚群的多态性、受选择区域等,进而挖掘出与群体性状相关基因区域,开发相关分子标记。
目前文章中常用的算法包括Fst分析(比较亚群分化程度)、Tajima’D分析(分析是否为中性进化或受正向选择)、pi分析(分析DNA多态性水平)等。常用软件Vcftools软件即可对这三种指标进行计算,进而分析群体间受选择的区域。
Q-17****:群体进化的实验设计思路?
A-17:材料选择:通常需要两个亚群以上,每个亚群选取10个样本左右(推荐动物≥10个,植物≥15个,珍稀物种可适当减少个体),总体建议不少于30个样本。
测序策略:目前基于Illumina Hiseq 4000平台进行双末端测序PE150测序。建议群体进化研究的测序深度不低于10X。
研究方法:一般群体进化的分析包括遗传多样性分析、基因交流情况分析、功能基因挖掘以及群体进化动态分析。
Q-18****:如果结题项目的结果文件中excel文件过大,如何打开?
A-18:可以使用UltraEdit、Editplus、Notepad++等文本编辑器打开。
Q-19****:GO分析样本如何来解读判断
A-19:Gene Ontology (简称GO):是一个国际化的基因功能分类体系,提供了一套动态更新的标准词汇来描述生物体中基因和基因产物的属性。包括3个Ontology分别描述基因分子功能(molecular function),所处的细胞位置(celluar component),参与的生物学过程(biological process)。
Q-20****:如何避免基因组中的重复序列造成的组装错误?
A-20:应用新一代高通量测序技术,构建170bp、500bp、2Kb、5Kb、10Kb、20Kb等不同大小的DNA测序文库,进行双末端大量测序,可以避免基因组中的重复序列造成的错拼。当测序数据量达到基因组大小的60倍以上时,即可保证基因组的完整性和序列中单碱基的准确性。
Q-21****:如何检测基因组组装的准确性?
A-21:目前,主要可以通过以下几种方法来检验基因组组装的准确性。
①通过构建BAC或Fosmid文库,并进行常规测序,将所得序列与拼接好的Contigs做比对以判断基因组组装的准确率。
②将已知的基因序列与拼接好的Scaffolds做比对,查看两者是否吻合,吻合度越高,表明基因组组装越好,而且已知的基因序列越多,评价结果越可靠。
③估计组装后基因组的单碱基准确度,利用新一代测序技术,如果95%以上的基因组单碱基覆盖度超过20×,则认为该基因组的单碱基准确度较高。