刘小泽写于18.8.21
争取用三部分整理完
Part I是前情提要,学习一些背景知识;
Part II是实战,从文章拿到数据开始,只要是服务器能完成的工作都完成;
Part III 将是R处理部分,也是最核心的部分,我会先学习Bioconductor,抓紧时间吧
. 阅读文献
文章梗概
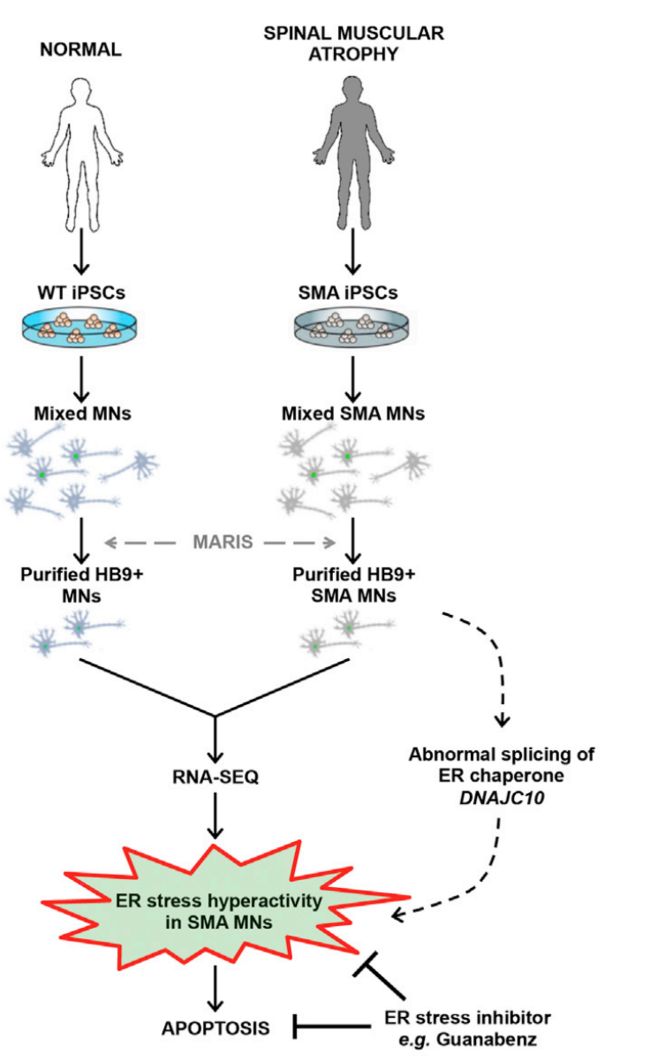
运动神经元存活蛋白(Survival Motor Neuron, SMN)是人体内的泛表达蛋白,它的会导致脊髓性肌萎速症(Spinal muscular atrophy, SMA),SMA是由SMN1基因突变引起的,但是这个基因是广泛表达的基因,为什么运动神经元(Motor Neurons, MNs)偏偏是最易受影响的细胞类型之一?实验通过对照组和SMA患者诱导多能干细胞(IPSC)从而产生纯化的运动神经元,通过固定、抗体标记,然后进行了RNA测序研究。在患者运动神经元中发现了SMA特异性的变化,其中包括内质网(ER)应激通路的过度激活。功能研究表明,抑制内质网的应激反应能提高运动神经元的存活率。在患病小鼠中,使用ER应激抑制剂穿过血脑屏障可以保护脊髓运动神经元。因此,选择性激活内质网应激反应,导致了SMA患病个体的运动神经元死亡。

开始实战
万事之源,软件为先
#配置conda
conda create -n rna-seq python=2 samtools fastp sra-tools hisat2 rseqc -y
conda install -c bcbio htseq -y
conda install numpy pysam -y
CONBIN=/home/biosoft/miniconda3/envs/rna-seq/bin
配置好工作路径
RNA=/home/project/rna-seq
mkdir -p $RNA/{raw_data,clean_data,ref/{genome,gtf,index},align,stats,matrix}
RAW=$RNA/raw_data
CLEAN=$RNA/clean-data
GENOME=$RNA/ref/genome
GTF=$RNA/ref/gtf
INDEX=$RNA/ref/index
ALIGN=$RNA/aign
MATRIX=$RNA/matrix
STATS=$RNA/stats
下载测试数据
GEO数据库中的编号是:GSE69175
关于数据:
NGS测序数据一般会上传到SRA数据库里面,但是怎么从GEO数据库中找到SRA原始数据的下载地址?【GEO数据库地址:https://www.ncbi.nlm.nih.gov/geo/】

具体的层级关系是:SRP(项目)—>SRS(样本)—>SRX(数据产生)—>SRR(数据本身)
SRR数据库地址:https://ftp-private.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/
##################################################################
################# Part 1: Data download #########################
##################################################################
# SRR2038215-SRR2038216: 对照
# SRR2038217-SRR2038218: SMN
cd $RAW
for i in `seq 15 18`;do
ascp -v -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh \
-k 1 -T -l200m [email protected]:/sra/sra-instant/reads/ByRun/sra/SRR/SRR358/SRR35899${i}/SRR35899${i}.sra ./ #使用ascp下载,嗖嗖嗖~
done
fastq-dump --gzip --split-3 *.sra #sra转为fastq
#以下3行原版来自生信技能树Jimmy
find $RAW -name "*.gz" | grep 1.fastq.gz >1
find $RAW -name "*.gz" | grep 2.fastq.gz >2
paste 1 2 > raw_conf #将read1和read2各自的合集再整合到一起,形成一个数据配置文件
cp raw_conf $CLEAN
质控过滤
这里使用的是fastp,同时融合了质控、过滤的模式;
同时也可以使用fastqc + trimmomatic/ trim_galore进行
##################################################################
################## Part 2: QC & trim ############################
##################################################################
source activate rna-seq
cd $CLEAN
#下面四行原版来自生信技能树Jimmy
cat raw_conf | while read id;do
line=(${id})
fq1=${line[0]}
fq2=${line[1]}
fastp -i $fq1 -I $fq2 -o out.$(basename $fq1) -O out.$(basename $fq2)-w 16
done
source deactivate
下载参考基因组和注释文件
人类参考基因组选择UCSC数据库;注释文件选择GENCODE,https://www.gencodegenes.org/
下载好基因组,需要构建基因组索引
如果自己的项目做非模式物种,可以用二代三代测序数据组装成参考转录组,例如trinity组装。如果做的物种有参考基因组,就方便一些,可以直接从相关数据库中下载参考基因组
##################################################################
############### Part 3: prepare ref & index ######################
##################################################################
##Download hg19
cd $GENOME
for i in $(seq 1 22) X Y M;do
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;
done
gunzip *.gz
for i in $(seq 1 22) X Y M;do
cat chr${i}.fa >> hg19.fa
done
rm chr*
##或者也可以直接下载成品
#wget http://10.10.0.8/hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz
##Download GRCh38 https://www.gencodegenes.org/releases/current.html
cd $GTF
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_28/GRCh37_mapping/gencode.v28lift37.annotation.gtf.gz
gunzip *.gz
## hisat2 index
cd $INDEX
hisat2-build -p 20 $GENOME/hg19.fa hg19 #-p为线程数
比对
##################################################################
######## Part 4: align & sort & index human samples:56-58 ########
##################################################################
source activate rna-seq
cd $ALIGN
for i in `seq 15 18`;do
hisat2 -t -p 20 -x $INDEX/hg19 \
-1 $CLEAN/out.SRR20382${i}_1.fastq.gz \
-2 $CLEAN/out.SRR20382${i}_2.fastq.gz \
-S SRR20382${i}.sam
samtools view -Sb SRR20382${i}.sam > SRR20382${i}.bam
samtools sort -@ 20 -o SRR20382${i}.sorted.bam SRR20382${i}.bam
samtools index SRR20382${i}.sorted.bam
rm *.sam
done
source deactivate
#关于排序:默认按染色体位置排序;-n根据read名排序;-t根据tag排序
使用了samtools的三件套:转换(view)、排序(sort)、建索引(index)
转换: -S指输入文件格式(不加-S默认输入是bam),-b指定输出文件(默认输出sam)【如果要bam转sam,-h设置输出sam时带上头注释信息】
排序: 对bam排序,-@ 线程数, -o输出文件
索引: 结果会产生.bai文件【必须排序后才能建索引~就像体育课必须从高到矮排好以后再报数】
基本信息统计
##################################################################
################ Part 5: basic statistics #######################
##################################################################
source activate rna-seq
cd $STATS
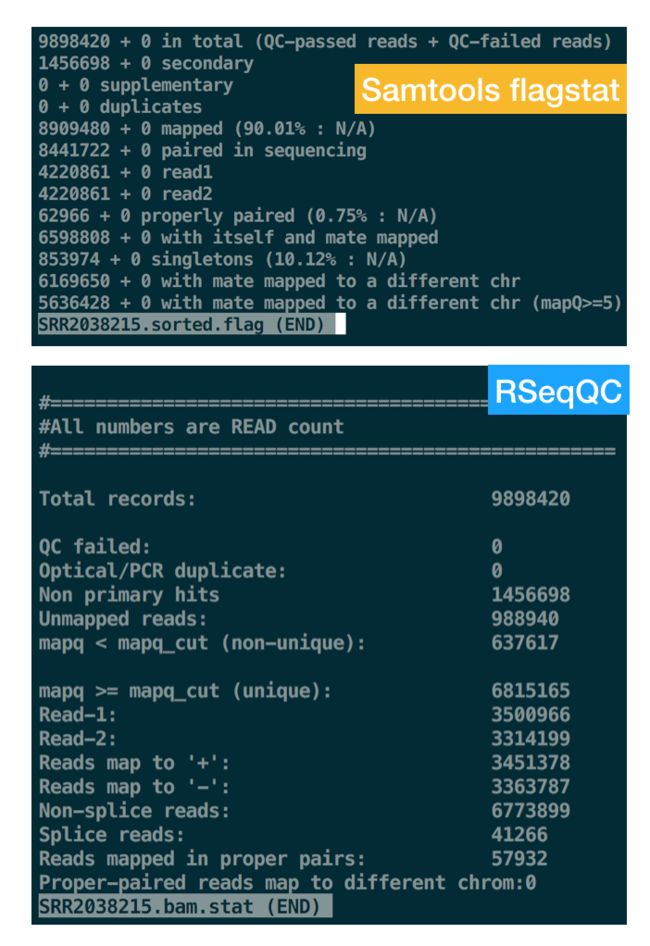
for i in `seq 15 18`;do
samtools flagstat $ALIGN/SRR20382${i}.sorted.bam > SRR20382${i}.sorted.flag
done
#如果想根据flag提取特定区域,比如想查看1号染色体100-10000的区域的信息
#samtools view -b -f 0x10 $ALIGN/SRR20382${i}.sorted.bam chr1:100-10000 > ${i}.flag.bam
#samtools flagstat ${i}.flag.bam
#使用RSeqQC统计
#先用bam_stat.py对bam文件统计,看下比对率
#bam_stat.py -i $ALIGN/SRR20382${i}.sorted.bam > SRR20382${i}.bam.stat
source deactivate
reads计数
基于基因组水平,可以用Htseq-count和featureCounts
-
Htseq-count:它是用python写的一个脚本,
conda install -c bcbio htseq -y安装好以后可以直接拿来用需要比对文件sam/bam格式;需要基因组注释文件gff/gtf格式。这两个文件的染色体名称必须相同,因为需要将比对位置和特征信息相匹配就要借助染色体名。
它的工作原理是:先通过bam文件找到reads比对上的外显子,然后去gtf文件中将外显子的基因ID进行统计,得到的结果就是未经标准化的表达量source activate rna-seq cd $MATRIX for i in `seq 15 18`;do $CONBIN/htseq-count -s no -r name -f bam $ALIGN/SRR20382${i}.sorted.bam \ $GTF/gencode.v28lift37.annotation.gtf \ >SRR20382${i}.count 2>SRR20382${i}.log done source deactivatehtseq-count的
-s参数是strand意思,默认使用链特异性建库方式,也就是计算过程中要求read1只能和双端测序的其中一条比较,read2只能和另一条比较;我们一般设置no即可,表示每条read都和正负链比较一次;
-r参数两个选择name和pos,数据根据位置或者read名称排序,默认name;
-f参数输入文件格式bam/sam;-m参数一般也就是默认union模式结果每个样本输出一个count文件,其中包含了基因名和reads计数;另外,如果你看到文件倒数几行,会发现还有几行带文字的
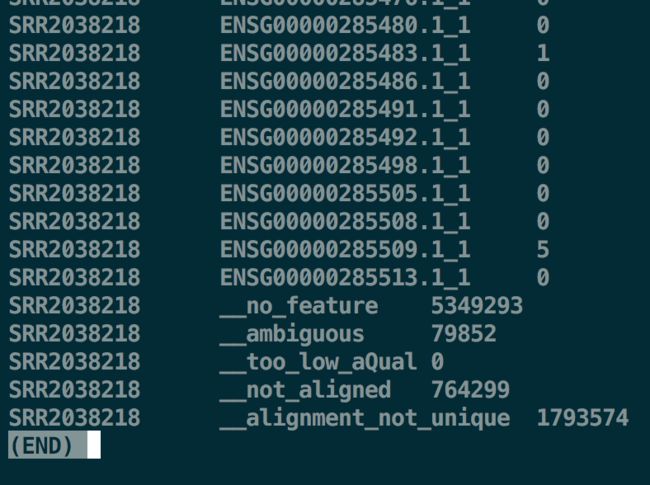 htseq-count计数
htseq-count计数no_feature:比对区域与任何基因都没有重叠
ambiguous:比对区域与多个基因都发生重叠
too_low_aQual:比对质量低于设定阈值(默认是10)
not_aligned:无法比对上
alignment_not_unique:比对位置不唯一 -
featureCounts:隶属于subread套件【相比于htseq更快】
source activate rna-seq cd $MATRIX begin=$(date +%s) for i in `seq 15 18`;do featureCounts -T 20 -p -t exon -g gene_id -a $GTF/gencode.v28lift37.annotation.gtf -o SRR20382${i}.feature.count $ALIGN/SRR20382${i}.bam done tim=$(echo "$(date +%s)-$begin" | bc) printf "time of featureCounts for 4 samples: %.4f seconds" $tim source deactivate #运行结果显示: #time of featureCounts for 4 samples: 366.5310 seconds-T参数设置线程,
-p参数表示针对双端测序数据,
-t 参数默认将外显子认定为基因组的一个feature(搜索特征),
-g参数默认按照基因名来计数,
-a参数输入注释文件,
-o参数输出文件结果有两个,一个count文件,一个summary文件
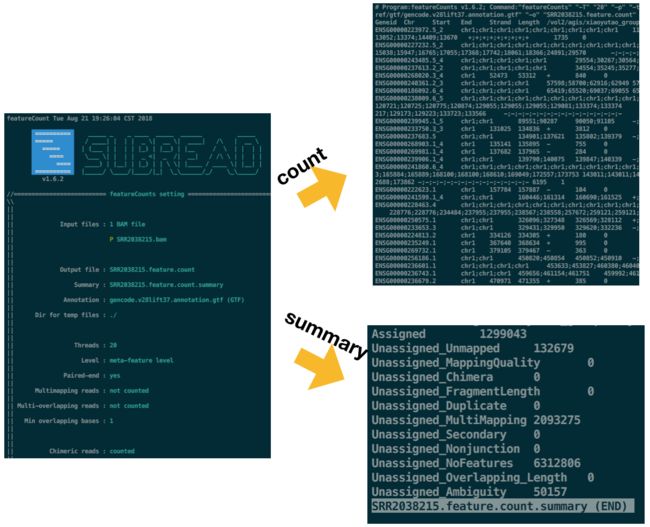 featureCounts文件
featureCounts文件
对两个软件的结果进行合并
##合并htdeq生成的count文件到matrix.count
cd $MATRIX/htseq
perl -lne 'if ($ARGV=~/(.*).count/){print "$1\t$_"}' *.count | grep -v "_" >matrix.count
##合并featureCounts生成的count文件到matrix_2.count
cd $MATRIX/featureCounts
for i in `seq 15 18`;do
cut -f 1,7 SRR20382${i}.feature.count | grep -v "^#" > SRR20382${i}.matrix
sed -i '1d' SRR20382${i}.matrix
done
perl -lne 'if ($ARGV=~/(.*).matrix/){print "$1\t$_"}' *.matrix >matrix_2.count
统计一下两个软件的计数之和
#统计featureCounts
perl -alne '$sum += $F[2]; END {print $sum}' matrix.count
#结果是5882943
#统计htseq-count,结果是786338
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
