任务
1.实现这个功能的软件也很多,还是烦请大家先自己搜索几个教程,入门请统一用htseq-count,对每个样本都会输出一个表达量文件。
2.需要用脚本合并所有的样本为表达矩阵。参考:生信编程直播第四题:多个同样的行列式文件合并起来。
3.对这个表达矩阵可以自己简单在excel或者R里面摸索,求平均值,方差。
看看一些生物学意义特殊的基因表现如何,比如GAPDH,β-ACTIN等等。
来源于生信技能树
下面摘自hoptop,想看更加详细的内容htseq的使用方法和计算原理点击这里
在上篇的比对中,我们需要纠结是否真的需要比对,如果你只需要知道已知基因的表达情况,那么可以选择alignment-free工具(例如salmon, sailfish),如果你需要找到noval isoforms,那么就需要alignment-based工具(如HISAT2, STAR)。到了这一篇的基因(转录本)定量,需要考虑的因素就更加多了,以至于我不知道如何说清才能理清逻辑。
定量分为三个水平
- 基因水平(gene-level)
- 转录本水平(transcript-level)
- 外显子使用水平(exon-usage-level)。
在基因水平上,常用的软件为HTSeq-count,featureCounts,BEDTools, Qualimap, Rsubread, GenomicRanges等。以常用的HTSeq-count为例,这些工具要解决的问题就是根据read和基因位置的overlap判断这个read到底是谁家的孩子。值得注意的是不同工具对multimapping reads处理方式也是不同的,例如HTSeq-count就直接当它们不存在。而Qualimpa则是一人一份,平均分配。
对每个基因计数之后得到的count matrix再后续的分析中,要注意标准化的问题。如果你要比较同一个样本(within-sample)不同基因之间的表达情况,你就需要考虑到转录本长度,因为转录本越长,那么检测的片段也会更多,直接比较等于让小孩和大人进行赛跑。如果你是比较不同样本(across sample)同一个基因的表达情况,虽然不必在意转录本长度,但是你要考虑到测序深度(sequence depth),毕竟测序深度越高,检测到的概率越大。除了这两个因素外,你还需要考虑GC%所导致的偏差,以及测序仪器的系统偏差。目前对read count标准化的算法有RPKM(SE), FPKM(PE),TPM, TMM等,不同算法之间的差异与换算方法已经有文章进行整理和吐槽了。但是,有一些下游分析的软件会要求是输入的count matrix是原始数据,未经标准化,比如说DESeq2,这个时候你需要注意你上一步所用软件会不会进行标准化。
在转录本水平上,一般常用工具为Cufflinks和它的继任者StringTie, eXpress。这些软件要处理的难题就时转录本亚型(isoforms)之间通常是有重叠的,当二代测序读长低于转录本长度时,如何进行区分?这些工具大多采用的都是expectation maximization(EM)。好在我们有三代测序。
上述软件都是alignment-based,目前许多alignment-free软件,如kallisto, silfish, salmon,能够省去比对这一步,直接得到read count,在运行效率上更高。不过最近一篇文献[1]指出这类方法在估计丰度时存在样本特异性和读长偏差。
在外显子使用水平上,其实和基因水平的统计类似。但是值得注意的是为了更好的计数,我们需要提供无重叠的外显子区域的gtf文件[2]。用于分析差异外显子使用的DEXSeq提供了一个Python脚本(dexseq_prepare_annotation.py)执行这个任务。小结
计数分为三个水平: gene-level, transcript-level, exon-usage-level
标准化方法: FPKM RPKM TMM TPM
htseq的使用方法和计算原理点击这里
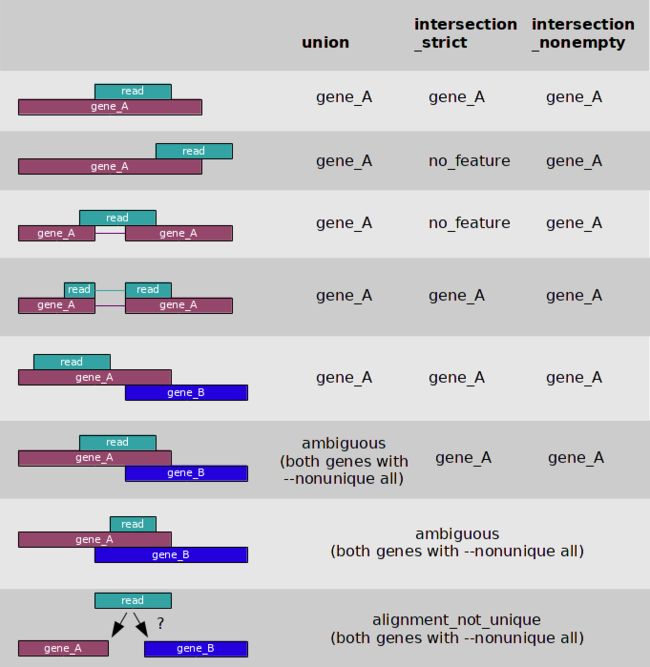
如何判断一个 reads 属于某个基因, htseq-count 提供了 union, intersection_strict,intersection_nonempty 3 种模型,如图(大多数情况下作者推荐用 union 模型),如果这三种模型还是不和你心意,可以通过htseq-count 自定义模型,方法详见 A tour through HTSeq 。
1.数据准备
输入为sam格式的文件,如果是paired-end(双端测序),必须对SAM文件按照reads名称排序(sort by name,not position)。对read name或 位置 进行排序皆可,通过 -r 可以指定您的数据是以什么方式排序的:
samtools sort -n accepted_hits_unique.bam -o accepted_hits_unique_name_sorted.bam # -n 按read name 排序 ,如果不指定则按染色体位置(pos)排序
具体来说:先用samtools先对bam文件排序,再转换为sam(这一步可以不要,不必进行转换)。请看Jimmy文章
# 首先将bam文件按reads名称进行排序(前期是按照默认的染色体位置进行排序的,所以要重新进行排序),这里我们主要以小鼠的数据为例子,不进行人类的测序数据。
$ cd /mnt/f/rna_seq/aligned
#人部分
# -n 按read name 排序 ,如果不指定则按染色体位置排序
$ for ((i=56;i<=58;i++));do samtools sort -n SRR35899${i}.bam -o SRR35899${i}_nsorted.bam;done
#小鼠部分
$ for ((i=59;i<=62;i++));do samtools sort -n SRR35899${i}.bam -o SRR35899${i}_nsorted.bam;done
# 将排序后的bam文件再次转换成sam格式的文件
#这一步可以省略不要
$ for ((i=56;i<=62;i++));do samtools view -h SRR35899${i}_nsorted.bam > SRR35899${i}_count.sam;done
2 reads计数,得到表达矩阵
数据准备已经完成,接下来要使用htseq-count对数据进行reads 计数。
Usage:htseq-count [options]
注:这里最好使用ensembl的基因组注释文件,小鼠注释文件下载地址。但是也可以用前面下载过的gencode注释文件。
安装htseq
# 安装htseq
$ conda install htseq
$ htseq-count --v
#usage: htseq-count [options] alignment_file gff_file
下载小鼠注释数据
看一下htseq-count帮助文件,了解下用法
$ htseq-count --h
(base) kelly@DESKTOP-MRA1M1F:/mnt/f/rna_seq/data/matrix$ htseq-count --h
usage: htseq-count [options] alignment_file gff_file
This script takes one or more alignment files in SAM/BAM format and a feature
file in GFF format and calculates for each feature the number of reads mapping
to it. See http://htseq.readthedocs.io/en/master/count.html for details.
positional arguments:
samfilenames Path to the SAM/BAM files containing the mapped reads.
If '-' is selected, read from standard input
featuresfilename Path to the file containing the features
optional arguments:
-h, --help show this help message and exit
-f {sam,bam}, --format {sam,bam}
type of data, either 'sam' or 'bam'
(default: sam)
-r {pos,name}, --order {pos,name}
'pos' or 'name'. Sorting order of
(default: name). Paired-end sequencing data must be
sorted either by position or by read name, and the
sorting order must be specified. Ignored for single-
end data.
可以看出,htseq支持SAM和BAM文件,所以就没必要把name-sorted BAM文件转成SAM文件,那样会用去很多时间和空间。这个通过-f bam解决。另外双端测序数据必须进行排序,看-r选项,即支持染色体位置排序(pos),又支持reads name排序,但name排序会更好。前面已经按name排序完成
#存放目录
$ cd mnt/f/rna_seq/data/reference/annotation/mm10
#下载(很快)
$ wget ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_mouse/release_M10/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf.gz
#解压
$ gunzip gencode.vM10.chr_patch_hapl_scaff.annotation.gtf.gz && rm -rf gencode.vM10.chr_patch_hapl_scaff.annotation.gtf.gz
#创建矩阵文件夹
$ cd /mnt/f/rna_seq/data && mkdir -p matrix && cd matrix
#使用htseq-count处理一个小鼠文件
$ htseq-count -r name -f bam /mnt/f/rna_seq/aligned/SRR3589959_nsorted.bam /mnt/f/rna_seq/data/reference/annotation/mm10/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf
还可以下面这样处理
for ((i=59;i<=62;i++));do htseq-count -r name -f bam /mnt/f/rna_seq/aligned/SRR35899${i}_nsorted.bam /mnt/f/rna_seq/data/reference/annotation/mm10/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf > /mnt/f/rna_seq/data/matrix/SRR35899${i}.count;done
或这样处理
for i in `seq 59 62`
do
htseq-count -r name -f bam /mnt/f/rna_seq/aligned/SRR35899${i}_nsorted.bam /mnt/f/rna_seq/data/reference/annotation/mm10/gencode.vM10.chr_patch_hapl_scaff.annotation.gtf > /mnt/f/rna_seq/data/matrix/SRR35899${i}.count 2> /mnt/f/rna_seq/data/matrix/SRR35899${i}.log
done
一共处理了4个小鼠的数据,大概5h,结果如下
(base) kelly@DESKTOP-MRA1M1F:/mnt/f/rna_seq/data/matrix$ ls -al *.count
-rwxrwxrwx 1 root root 1164655 Aug 1 12:35 SRR3589959.count
-rwxrwxrwx 1 root root 1168522 Aug 1 14:09 SRR3589960.count
-rwxrwxrwx 1 root root 1165311 Aug 1 15:17 SRR3589961.count
-rwxrwxrwx 1 root root 1166505 Aug 1 16:39 SRR3589962.count
- 对结果进行统计
$ wc -l *.count
48825 SRR3589959.count
48825 SRR3589960.count
48825 SRR3589961.count
48825 SRR3589962.count
195300 total
- 看下每个文件的格式,查看前4行,第一列ensembl_gene_id,第二列read_count计数
$ head -n 4 *.count
==> SRR3589959.count <==
ENSMUSG00000000001.4 1648
ENSMUSG00000000003.15 0
ENSMUSG00000000028.14 835
ENSMUSG00000000031.15 65
==> SRR3589960.count <==
ENSMUSG00000000001.4 2941
ENSMUSG00000000003.15 0
ENSMUSG00000000028.14 1366
ENSMUSG00000000031.15 52
==> SRR3589961.count <==
ENSMUSG00000000001.4 2306
ENSMUSG00000000003.15 0
ENSMUSG00000000028.14 950
ENSMUSG00000000031.15 83
==> SRR3589962.count <==
ENSMUSG00000000001.4 2780
ENSMUSG00000000003.15 0
ENSMUSG00000000028.14 1051
ENSMUSG00000000031.15 53
- 后4行
$ tail -n 4 *.count
==> SRR3589959.count <==
__ambiguous 295498
__too_low_aQual 1107674
__not_aligned 1025857
__alignment_not_unique 11278964
==> SRR3589960.count <==
__ambiguous 498973
__too_low_aQual 1799176
__not_aligned 1675660
__alignment_not_unique 18440618
==> SRR3589961.count <==
__ambiguous 425708
__too_low_aQual 1303141
__not_aligned 1067038
__alignment_not_unique 14080481
==> SRR3589962.count <==
__ambiguous 381237
__too_low_aQual 1542845
__not_aligned 1529309
__alignment_not_unique 14495860
3 合并表达矩阵并进行注释(R中进行)
-
从上面看出需要至少做两步工作才能更好理解和往下进行分析
第一,需要把4个文件合并;
第二,需要把ensembl_gene_id转换为gene_symbol;(这一步不进行也行,后面还需要)
所以,上一步得到的4个单独的矩阵文件,现在要把这4个文件合并为行为基因名,列为样本名,中间为count的矩阵文件。
- 熟悉python的朋友可以参考这篇文章
- 我用R中的merge命令来处理,参考这里和这里
先启动R_studio
(1) 载入数据,添加列名
再看下原始数据,可见59和61和control,60和62是实验
> options(stringsAsFactors = FALSE)
> control1<-read.table("SRR3589959.count",sep = "\t",col.names = c("gene_id","control1"))
> head(control1)
gene_id control1
1 ENSMUSG00000000001.4 1648
2 ENSMUSG00000000003.15 0
3 ENSMUSG00000000028.14 835
4 ENSMUSG00000000031.15 65
5 ENSMUSG00000000037.16 70
6 ENSMUSG00000000049.11 0
> control2<-read.table("SRR3589961.count",sep = "\t",col.names = c("gene_id","control2"))
> treat1<-read.table("SRR3589960.count",sep = "\t",col.names = c("gene_id","treat1"))
> treat2<-read.table("SRR3589962.count",sep = "\t",col.names = c("gene_id","treat2"))
(2)数据整合
-
merge进行整合 -
gencode的注释文件中的gene_id(如ENSMUSG00000105298.13_3)在EBI是不能搜索到的,所以用
gsub功能只保留ENSMUSG00000105298这部分
处理之前先看一下,也就是最好5行是我们不需要的,可以删除
> tail(control1)
gene_id control1
48820 ENSMUSG00000110424.1 26
48821 __no_feature 12642161
48822 __ambiguous 295498
48823 __too_low_aQual 1107674
48824 __not_aligned 1025857
48825 __alignment_not_unique 11278964
当然上面这个命令也可以在linux下执行
$ tail -n 10 *.count
进行整合
> raw_count <- merge(merge(control1, control2, by="gene_id"), merge(treat1, treat2, by="gene_id"))
> head(raw_count)
gene_id control1 control2 treat1 treat2
1 __alignment_not_unique 11278964 14080481 18440618 14495860
2 __ambiguous 295498 425708 498973 381237
3 __no_feature 12642161 15042888 22357626 18675857
4 __not_aligned 1025857 1067038 1675660 1529309
5 __too_low_aQual 1107674 1303141 1799176 1542845
6 ENSMUSG00000000001.4 1648 2306 2941 2780
> tail(raw_count)
gene_id control1 control2 treat1 treat2
48820 ENSMUSG00000110419.1 46 39 68 58
48821 ENSMUSG00000110420.1 0 0 0 0
48822 ENSMUSG00000110421.1 0 0 0 1
48823 ENSMUSG00000110422.1 1 0 1 2
48824 ENSMUSG00000110423.1 0 0 0 0
48825 ENSMUSG00000110424.1 26 20 48 24
删除前5行
#这里要注意,我读入之后顺序变了,删除的时候看下删除的是哪些行
> raw_count_filt <- raw_count[-1:-5,]
gene_id control1 control2 treat1 treat2
6 ENSMUSG00000000001.4 1648 2306 2941 2780
7 ENSMUSG00000000003.15 0 0 0 0
8 ENSMUSG00000000028.14 835 950 1366 1051
9 ENSMUSG00000000031.15 65 83 52 53
10 ENSMUSG00000000037.16 70 53 94 66
11 ENSMUSG00000000049.11 0 3 4 5
因为我们无法在EBI数据库上直接搜索找到ENSMUSG00000024045.5这样的基因,只能是ENSMUSG00000024045的整数,没有小数点,所以需要进一步替换为整数的形式。
# 第一步将匹配到的.以及后面的数字连续匹配并替换为空,并赋值给ENSEMBL
>ENSEMBL <- gsub("\\.\\d*", "", raw_count_filt$gene_id)
# 将ENSEMBL重新添加到raw_count_filt1矩阵
>row.names(raw_count_filt) <- ENSEMBL
>head(raw_count_filt)
gene_id control1 control2 treat1 treat2
ENSMUSG00000000001 ENSMUSG00000000001.4 1648 2306 2941 2780
ENSMUSG00000000003 ENSMUSG00000000003.15 0 0 0 0
ENSMUSG00000000028 ENSMUSG00000000028.14 835 950 1366 1051
ENSMUSG00000000031 ENSMUSG00000000031.15 65 83 52 53
ENSMUSG00000000037 ENSMUSG00000000037.16 70 53 94 66
ENSMUSG00000000049 ENSMUSG00000000049.11 0 3 4 5
(3)对基因进行注释-获取gene_symbol
以下两种方式可以进行
第一:去这里或这里的网页版,输入列表即可输出,不再赘述
第二:用bioMart对ensembl_id转换成gene_symbol
> library('biomaRt')
> library("curl")
Warning message:
package ‘curl’ was built under R version 3.4.4
> mart <- useDataset("mmusculus_gene_ensembl", useMart("ensembl"))
> my_ensembl_gene_id<-row.names(raw_count_filt)
> #Sys.setenv(https_proxy="http://proxy:8000")
> options(timeout = 4000000)
> my_ensembl_gene_id<-row.names(raw_count_filt)
> mms_symbols<- getBM(attributes=c('ensembl_gene_id','hgnc_symbol',"chromosome_name", "start_position","end_position", "band"),
+ filters = 'ensembl_gene_id', values = my_ensembl_gene_id, mart = mart)
#于是,不幸总是出错,我还是用了网页版工具进行转换
> readcount<-read.csv(file="raw_count_filt.csv",header = TRUE)
> head(readcount)
ensembl_id gene_id gene_symbol control1 control2 treat1 treat2
1 ENSMUSG00000000001 ENSMUSG00000000001.4 Gnai3 1648 2306 2941 2780
2 ENSMUSG00000000003 ENSMUSG00000000003.15 Pbsn 0 0 0 0
3 ENSMUSG00000000028 ENSMUSG00000000028.14 Cdc45 835 950 1366 1051
4 ENSMUSG00000000031 ENSMUSG00000000031.15 H19 65 83 52 53
5 ENSMUSG00000000037 ENSMUSG00000000037.16 Scml2 70 53 94 66
6 ENSMUSG00000000049 ENSMUSG00000000049.11 Apoh 0 3 4 5
#查看Akap8(别名AKAP95)表达情况
> Akap8 <- readcount[readcount$gene_symbol=="Akap8",]
> Akap8
ensembl_id gene_id gene_symbol control1 control2 treat1 treat2
4265 ENSMUSG00000024045 ENSMUSG00000024045.5 Akap8 936 1368 3386 4116
#treat中显著升高
---------------说明------------------------
以前用bioMart包的getBM命令没任何问题,这次却在这里停留了半天了。一直出下面的错误提示,(你们运行上面代码很可能不会出错)
Batch submitting query [===================================>-------------------------------] 53% eta: 4mError in curl::curl_fetch_memory(url, handle = handle) :
Timeout was reached: Connection timed out after 10000 milliseconds
于是不断google,尝试解决,尝试了以下方法:
- 1 可能是代理问题,所以采取直接连接和代理连接,未果
- 2 添加
Sys.setenv(https_proxy="http://proxy:8000"),未果 - 3 添加
options(timeout = 4000000), 未果
至此,我已经快崩溃了,不想在这一步一直停留,因为这并不影响下一步
又找到一个方法 - 4 把https://www.esemble.org添加到安装站点,未果
==================================
后记
经过RNA-seq(7): 筛选差异表达基因(DEGs)部分分析可知,上面代码没有问题,估计是因为我的网实在太不好,再加上行数太多(4万多行)。
==================================
4 输出count矩阵文件
readcount<-raw_count_filter[ ,-1]
write.csv(readcount, file='readcount.csv')