Hemberg-lab单细胞转录组数据分析(一)
Hemberg-lab单细胞转录组数据分析(二)
Hemberg-lab单细胞转录组数据分析(三)
Hemberg-lab单细胞转录组数据分析(四)
Hemberg-lab单细胞转录组数据分析(五)
收藏|北大生信平台"单细胞分析、染色质分析"视频和PPT分享
该如何自学入门生物信息学
生物信息之程序学习
构建表达矩阵
scRNA-seq数据的许多分析以表达矩阵为起点。一般来讲,表达矩阵的每一行代表一个基因,每一列代表一个细胞(但是一些作者会做个转置)。每个条目代表特定基因在给定细胞中的表达水平。而表达值的测量单位取决于建库方案和所用的标准化方法。
reads质控
见前面章节FastQC部分。
另外,使用Integrative Genomics Browser(IGV)或SeqMonk通常对数据可视化很有帮助,具体见下。
测序数据可视化 (一)
IGV基因组浏览器可视化高通量测序数据
高通量数据分析必备-基因组浏览器使用介绍 - 1
高通量数据分析必备-基因组浏览器使用介绍 - 2
高通量数据分析必备-基因组浏览器使用介绍 - 3
Reads比对
见前面章节STAR部分和Kallisto部分。
注释的很好的模式生物(例如小鼠,人)有着大量全长转录本数据集,伪比对方法(例如Kallisto,Salmon)可能优于常规比对方法。drop-seq方法获得的数据集有数以千万条reads,伪比对工具的运行时间比传统比对工具会少几个数量级,更有时间优势。从39个转录组分析工具,120种组合评估(转录组分析工具哪家强-导读版)一文中可以看出,伪比对工具的准确性和稳定性也相对比较高。
用STAR比对的操作示例 (前面章节部分更详细)
STAR --runThreadN 1 --runMode alignReads
--readFilesIn reads1.fq.gz reads2.fq.gz --readFilesCommand zcat --genomeDir
--parametersFiles FileOfMoreParameters.txt --outFileNamePrefix /output
注意,如果用了spike-ins(已知浓度的外源RNA分子),在比对前应该将参考基因组和spike-in分子的DNA序列合并作为共同”参考基因组”。具体见之前文件格式部分。
注意,使用UMI时,应从read序列中删除其条形码。常见的是将条形码加到read名称上。
一旦reads完成了到基因组的比对,我们需要检查比对率和确保有足够多的reads比对回了参考基因组。根据我们的经验,小鼠或人类细胞中read的比对率为60-70%。但是这个结果可能会因建库方案、read长度和比对工具参数设置而有所不同。常规上,我们希望所有细胞都具有相似的read比对率。如果有样品比对率异常低或比对回去的reads异常低,则需要多加注意甚至从后续分析中移除。较低的read比对率通常表示存在污染。生信宝典建议取出几十条未必对回去的reads做个blast,看下能否比对到其它物种来确定是污染还是测序错误还是比对参数设置的问题。
一个用Salmon量化表达操作的例子
salmon quant -i salmon_transcript_index -1 reads1.fq.gz -2 reads2.fq.gz -p #threads -l A -g genome.gtf --seqBias --gcBias --posBias
注意 Salmon操作会得到一个估计的read counts和transcripts per million(tpm)。根据我们的经验,TPM对单细胞测序中长基因的表达做了过度校正,因此我们建议使用read counts。
比对示例
下面直方图 (http://www.ehbio.com/ImageGP)显示了scRNA-seq实验中每个细胞不同比对状态的reads的数目。每个柱子代表一个细胞,按细胞的总read数升序排列。三个红色箭头标记的是比对到基因组的reads较低的异常样本,应该在后续分析中移除。两个黄色箭头指的是unmapped reads数目十分大的细胞。该例中,在比对质控期间这两个细胞会保留下来,但后期细胞质控时这两个细胞会因为核糖体RNA reads比例过高而移除。

Mapping QC
在把原始序列比对到基因组后,需要评估比对质量。这可以从多个角度进行评估,包括:rRNA/tRNAs的reads的占比或总量,reads在基因组上唯一比对位置的比例,比对到splice junction的reads比例,reads在转录本的覆盖均一性或深度。而为Bulk RNA-seq开发的方法如RSeQC,也适用于单细胞数据:
#pip install RSeQC
geneBody_coverage.py -i input.bam -r genome.bed -o output.txt
bam_stat.py -i input.bam -r genome.bed -o output.txt
split_bam.py -i input.bam -r rRNAmask.bed -o output.txt
然而,预期结果的评估取决于采用的建库方案,例如许多scRNA-seq用poly-A selection捕获转录本。这个方法可以排除核糖体RNA污染,但会导致3'区域更容易测到。下图展示了测序reads分布的3'偏好性,和去除的三个异常细胞的结果 (应该是最下面3条,推测是降解严重)。

Reads量化
scRNA-seq基因定量计算可以用bulk RNA-seq一样的工具,比如HT-seq or FeatureCounts。
# include multimapping
featureCounts -O -M -Q 30 -p -a genome.gtf -o outputfile input.bam
# exclude multimapping
featureCounts -Q 30 -p -a genome.gtf -o outputfile input.bam
唯一分子标识符UMI让计算转录本的绝对量成为可能,并且在scRNA-seq中很受欢迎。我们将在下一章讨论如何处理UMI。
唯一分子标识符(Unique Molecular Identifiers, UMI)
感谢EMBL Monterotondo的 Andreas Buness 在本节的合作。
UMI添加到每个转录本上
唯一分子标识符 (UMI)是在反转录过程中添加到转录本上的短的(4-10 bp)随机条形码序列。它们使得测序reads可以对应到单个转录本,从而去除扩增噪声和偏好性。

当测序含UMI的文库时,仅对包含UMI的转录本末端 (通常为3'末端)进行性测序。
比对UMI条形码
由于UMI数量(, N是UMIs的长度值)比每个细胞中的RNA分子数(~)少得多,每个UMI条形码可能会连接到多个转录本,因此需要借助条形码序列和reads比对位置两个条件鉴定起始的转录本分子。第一步是比对UMI reads,推荐用STAR来处理,因为它处理速度快且输出高质量的BAM比对。此外,比对位置的准确性对识别新的3'UTR区域很有意义。
UMI测序通常由双端reads组成,其中一端read是捕获细胞和UMI的条形码,而另一端read包含转录本的外显子序列。注意,推荐去除reads中的poly-A序列部分,以免这些reads比对到转录本内部poly-A或poly-T序列而产生错误。
处理UMI实验中的reads,通常有以下惯例:
UMI被添加到另一个配对read的序列名称中。
reads按细胞条形码分类到单独的文件中 (见前面的文章)。但对于细胞量极大的低深度测序数据集 (drop-seq),可以将细胞条形码添加到read名称中而不是拆分为单独文件以减少文件数量。

Counting 条形码
理论上,每个唯一的UMI-转录本对应该对应来源于一个RNA分子的所有reads。但是现实往往并非如此,最常见的原因是:
不同的UMI序列不一定表示它们是不同的UMI分子由于PCR或测序错误,碱基替换可能导致新的
UMI序列。较长的UMI出现碱基替换的机会更多。根据细胞条码测序错误估计,7-10%的10 bp长度的UMI中至少有一个碱基替换。如果错误没有纠正,将会过高估计转录本的数目。不同的转录本不一定是不同的分子比对错误或多个比对位置可能导致某些
UMI对应到错误的基因/转录本。这种类型的错误也会导致过高估计转录本的数目。相同的UMI不一定意味着相同的分子UMI频次的不同和短UMI可导致同一UMI和相同基因的不同mRNA分子相连,进而可能低估转录本数量。

错误纠正
如何最好的校正UMIs中的这些问题仍然是一个活跃的研究领域。我们自己认为的最好的解决上述问题的方法是:
UMI-tools’,设计了
directional-adjacency算法,同时考虑错配数目和相似UMI的相对频率来识别PCR和测序错误。(alevin, cellranger都是不错的选择,后面详细介绍)问题还无法完全解决,但通过删除只有很少read支持的
UMIs-转录本对,或者移除所有多比对位置的reads,可能会减轻该问题。Simple saturation (也称为”collision probability”)方法来估计分子的数量

其中N=唯一UMI条形码的总数,n=观察到的条形码数。
这个方法的一个重要缺陷是它假设所有UMI出现频率相同。但因为序列GC含量不同引入的偏差使得这一假设在大多数情况下这是不正确的。
如何最好地处理和使用UMI在目前生物信息学界是一个活跃的研究领域。而我们了解到的几种最近开发的方法有:
UMI-tools
PoissonUMIs
zUMIs
dropEst
下游分析
当前的UMI平台(DropSeq,InDrop,ICell8)展现出从低到高变化很大的捕获效率,如下图所示。

这一高可变性可能会引入很强的偏差,需要在下游分析时考虑到。现在的分析通常根据细胞类型或生物通路把细胞/gene混合一起增加检测能力。更合适的统计分析方法亟待研究以便更好地调整这些偏差,使得结果更能反映真实现象。
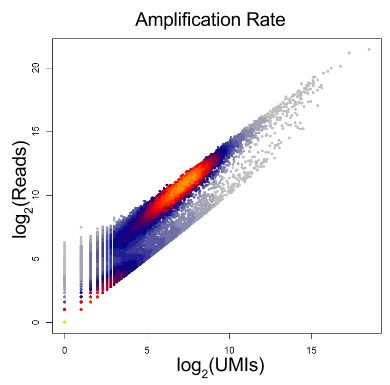
练习1 数据是三个不同来源的诱导多功能干细胞的UMI counts和read counts (有关此数据集的详细信息请参阅后续文章)。
umi_counts <- read.table("tung/molecules.txt", sep = "\t")
read_counts <- read.table("tung/reads.txt", sep = "\t")
使用此数据:
绘制捕获效率的变化
确定扩增率:每个UMI对应的平均reads数。
# Exercise 1
# Part 1
plot(colSums(umi_counts), colSums(umi_counts > 0), xlab="Total Molecules Detected", ylab="Total Genes Detected")
# Part 2
amp_rate <- sum(read_counts)/sum(umi_counts)
amp_rate