上期我们介绍了Swiss-model进行同源建模,虽然建模自动化非常方便,但是自己可以调节的地方较少,对待序列相似度较高的还不错,但是对待序列相似度低的话可能建模的质量就不怎么好了,所以今天给大家介绍同源建模界的鼻祖同时也是同源建模的大哥大,Modeller,要知道我们常用的商业软件Discover Studio以及YASARA,其同源建模实际上就是使用的Modeller。这也侧面说明了Modeller在同源建模界的地位。
由于Modeller是命令化使用,所以有许多人对其心存敬畏,但是真正接触后可以发现其在强大的基础上的平易近人,今天就带领大家初识Modeller:
软件下载与简单安装
下载地址: https://salilab.org/modeller/download_installation.html
Modeller 的license: MODELIRANJE
注意,该软件仅学术用户免费
Window安装
双击解压32位或者64位文件即可
Linux安装(Debian/Ubuntu)
sudo env KEY_MODELLER=MODELIRANJE dpkg - i 包名称.deb
Modeller 之单模板建模
Modeller虽然自带搜索模板功能,但是其每次更新数据库比较麻烦,还是建议用NCBI BLAST进行搜索,首先打开NCBI,输入蛋白名称:
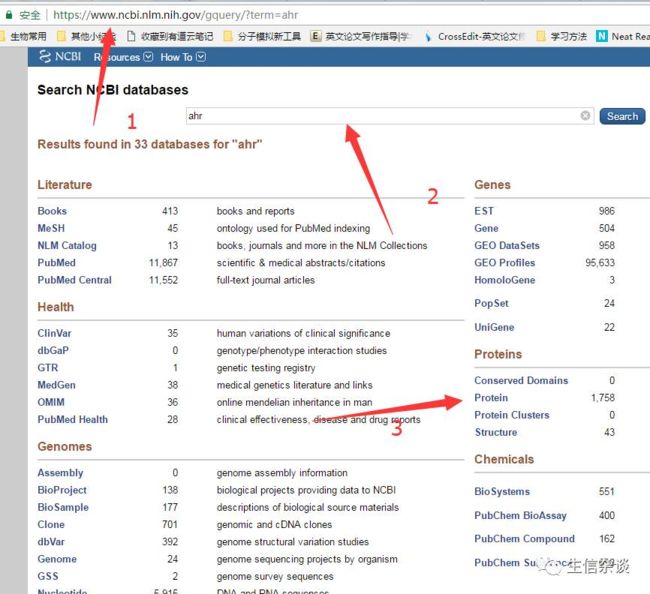
点击蛋白分类,右侧选择种属,然后选择自己的目标序列。
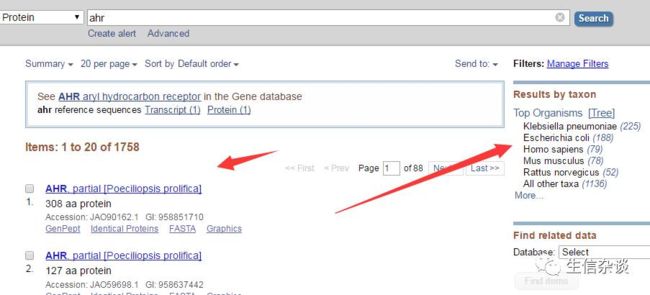
再点击右侧的RUN BLAST,在BLAST 里选择搜索的库位pdb数据库,后blast。

步骤原理与Swiss-Model类似,结果选择的方式与Swiss-Model相似,由于我们进行的是单模板建模,所以选择一个质量最好的已知晶体结构进行建模。(下次教程会提到多模板建模)。同时下载目标序列。将目标序列修改成如下:
1.修改目标序列
>P1;序列名称
sequence:目标序列名称:::::::0.00: 0.00
此处填写自己的序列*
File: 目标序列名称.ali
例子:
>P1;TvLDH
sequence:TvLDH:::::::0.00: 0.00
MSEAAHVLITGAAGQIGYILSHWIASGELYGDRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLAGFVATTDPKAAFKDIDCAFLVASMPLKPGQVRADLISSNSVIFKNTGEYLSKWAKPSVKVLVIGNPDNTNCEIAMLHAKNLKPENFSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGESMVADLTQATFTKEGKTQKVVDVLDHDYVFDTFFKKIGHRAWDILEHRGFTSAASPTKAAIQHMKAWLFGTAPGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVDKEGKIHVVEGFKVNDWLREKLDFTEKDLFHEKEIALNHLAQGG*
File: TvLDH.ali
其中>P1;序列名称写在第一行,为索引头,第二列的sequence:表示文件包含未知结构的序列,*表示结尾标记
2.目标序列与模板进行比对
from modeller import *
env = environ()
aln = alignment(env)
mdl = model(env, file='模板文件名', model_segment=('FIRST:链','LAST:链'))
aln.append_model(mdl, align_codes='模板pdb名称+链', atom_files='模板文件名.pdb')
aln.append(file='目标序列名称.ali', align_codes='目标序列名称')
aln.align2d()
aln.write(file='保存文件名称.ali', alignment_format='PIR')
aln.write(file='保存文件名称.pap', alignment_format='PAP')
File:脚本名称1.py
例子:
from modeller import *
env = environ()
aln = alignment(env)
mdl = model(env, file='1bdm', model_segment=('FIRST:A','LAST:A'))
aln.append_model(mdl, align_codes='1bdmA', atom_files='1bdm.pdb')
aln.append(file='TvLDH.ali', align_codes='TvLDH')
aln.align2d()
aln.write(file='TvLDH-1bdmA.ali', alignment_format='PIR')
aln.write(file='TvLDH-1bdmA.pap', alignment_format='PAP')
File: align2d.py
首先我们在python中导入modeller模块,然后创建一个environ()的实例,再然后创建一个空的比对aln模型。再创建一个新的蛋白模型,mdl,该蛋白模型是保存了的模板序列的优化。append_model()命令转化模型的pdb序列进入aln内,append()命令将目标序列同样也加入。执行align2d()实例进行比对。然后写出比对的结果为PIR与PAP格式。
使用modeller执行脚本命令
mod+版本号 脚本1.py
3.构建模型
from modeller import *
from modeller.automodel import *
#from modeller import soap_protein_od
env = environ()
a = automodel(env, alnfile='比对好的文件.ali',
knowns='已知模板+链', sequence='目标序列',
assess_methods=(assess.DOPE,
#soap_protein_od.Scorer(),
assess.GA341))
a.starting_model = 模型其实编号
a.ending_model = 模型结束编号
a.make()
File: 脚本名称2.py
例子
from modeller import *
from modeller.automodel import *
#from modeller import soap_protein_od
env = environ()
a = automodel(env, alnfile='TvLDH-1bdmA.ali',
knowns='1bdmA', sequence='TvLDH',
assess_methods=(assess.DOPE,
#soap_protein_od.Scorer(),
assess.GA341))
a.starting_model = 1
a.ending_model = 5
a.make()
采用自动建模实例,automodel(),并选择已知模型,目标序列,assess_methods()内选择模型的评价方法,具体的评价方法后头将会再讲。make()实例完成建模的计算。
使用modeller执行脚本命令
mod+版本号 脚本1.py
完成后对应的脚本1.log将会产生日志文件,里面有用的模型评价整体内容。
例子:
>> Summary of successfully produced models:
Filename molpdf DOPE score GA341 score
----------------------------------------------------------------------
TvLDH.B99990001.pdb 1763.56104 -38079.76172 1.00000
TvLDH.B99990002.pdb 1560.93396 -38515.98047 1.00000
TvLDH.B99990003.pdb 1712.44104 -37984.30859 1.00000
TvLDH.B99990004.pdb 1720.70801 -37869.91406 1.00000
TvLDH.B99990005.pdb 1840.91772 -38052.00781 1.00000
4.模型评价
from modeller import *
from modeller.scripts import complete_pdb
log.verbose() # request verbose output
env = environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology
env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# read model file
mdl = complete_pdb(env, '评价的模型.pdb')
# Assess with DOPE:
s = selection(mdl) # all atom selection
s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='文件名称.profile',
normalize_profile=True, smoothing_window=15)
File: 脚本3.py
使用complete_pdb脚本对PDB文件进行读取并准备其的能力计算,DOPE能量计算使用assess_dope命令,每个滑块窗使用15个残基(ERRAT2为9个),得出的文件名称.profile可以用GNUPLOT或者例如python的matplotlib包进行读取。
结果例子如下:
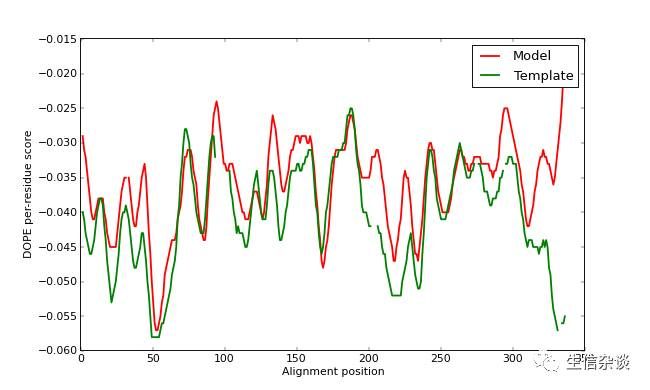
可以看出序列220-250结果没有模板模型质量的好,可以进行下一步的loop 优化,我们下期详细讲解,敬请关注。
更多原创精彩内容敬请关注生信杂谈**
